Piero Ruscitti, Luca Cantarini, Peter A. Nigrovic, Dennis McGonagle, Roberto Giacomelli
{"title":"斯蒂尔病的最新进展和不断演变的概念","authors":"Piero Ruscitti, Luca Cantarini, Peter A. Nigrovic, Dennis McGonagle, Roberto Giacomelli","doi":"10.1038/s41584-023-01065-6","DOIUrl":null,"url":null,"abstract":"Still’s disease is a rare inflammatory syndrome that encompasses systemic juvenile idiopathic arthritis and adult-onset Still’s disease, both of which can exhibit life-threatening complications, including macrophage activation syndrome (MAS), a secondary form of haemophagocytic lymphohistiocytosis. Genetic insights into Still’s disease involve both HLA and non-HLA susceptibility genes, suggesting the involvement of adaptive immune cell-mediated immunity. At the same time, phenotypic evidence indicates the involvement of autoinflammatory processes. Evidence also implicates the type I interferon signature, mechanistic target of rapamycin complex 1 signalling and ferritin in the pathogenesis of Still’s disease and MAS. Pathological entities associated with Still’s disease include lung disease that could be associated with biologic DMARDs and with the occurrence of MAS. Historically, monophasic, recurrent and persistent Still’s disease courses were recognized. Newer proposals of alternative Still’s disease clusters could enable better dissection of clinical heterogeneity on the basis of immune cell profiles that could represent diverse endotypes or phases of disease activity. Therapeutically, data on IL-1 and IL-6 antagonism and Janus kinase inhibition suggest the importance of early administration in Still’s disease. Furthermore, there is evidence that patients who develop MAS can be treated with IFNγ antagonism. Despite these developments, unmet needs remain that can form the basis for the design of future studies leading to improvement of disease management. In this Review, the authors describe shared pathophysiology of systemic juvenile idiopathic arthritis and adult-onset Still’s disease and their life-threatening complication, macrophage activation syndrome. Therapeutic developments now enable the targeting of multiple pathways in these conditions, and evidence suggests that early use of DMARDs has the potential to prevent chronic disease.","PeriodicalId":18810,"journal":{"name":"Nature Reviews Rheumatology","volume":"20 2","pages":"116-132"},"PeriodicalIF":32.7000,"publicationDate":"2024-01-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Recent advances and evolving concepts in Still’s disease\",\"authors\":\"Piero Ruscitti, Luca Cantarini, Peter A. Nigrovic, Dennis McGonagle, Roberto Giacomelli\",\"doi\":\"10.1038/s41584-023-01065-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Still’s disease is a rare inflammatory syndrome that encompasses systemic juvenile idiopathic arthritis and adult-onset Still’s disease, both of which can exhibit life-threatening complications, including macrophage activation syndrome (MAS), a secondary form of haemophagocytic lymphohistiocytosis. Genetic insights into Still’s disease involve both HLA and non-HLA susceptibility genes, suggesting the involvement of adaptive immune cell-mediated immunity. At the same time, phenotypic evidence indicates the involvement of autoinflammatory processes. Evidence also implicates the type I interferon signature, mechanistic target of rapamycin complex 1 signalling and ferritin in the pathogenesis of Still’s disease and MAS. Pathological entities associated with Still’s disease include lung disease that could be associated with biologic DMARDs and with the occurrence of MAS. Historically, monophasic, recurrent and persistent Still’s disease courses were recognized. Newer proposals of alternative Still’s disease clusters could enable better dissection of clinical heterogeneity on the basis of immune cell profiles that could represent diverse endotypes or phases of disease activity. Therapeutically, data on IL-1 and IL-6 antagonism and Janus kinase inhibition suggest the importance of early administration in Still’s disease. Furthermore, there is evidence that patients who develop MAS can be treated with IFNγ antagonism. Despite these developments, unmet needs remain that can form the basis for the design of future studies leading to improvement of disease management. In this Review, the authors describe shared pathophysiology of systemic juvenile idiopathic arthritis and adult-onset Still’s disease and their life-threatening complication, macrophage activation syndrome. Therapeutic developments now enable the targeting of multiple pathways in these conditions, and evidence suggests that early use of DMARDs has the potential to prevent chronic disease.\",\"PeriodicalId\":18810,\"journal\":{\"name\":\"Nature Reviews Rheumatology\",\"volume\":\"20 2\",\"pages\":\"116-132\"},\"PeriodicalIF\":32.7000,\"publicationDate\":\"2024-01-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Reviews Rheumatology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.nature.com/articles/s41584-023-01065-6\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"RHEUMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Reviews Rheumatology","FirstCategoryId":"3","ListUrlMain":"https://www.nature.com/articles/s41584-023-01065-6","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"RHEUMATOLOGY","Score":null,"Total":0}
Recent advances and evolving concepts in Still’s disease
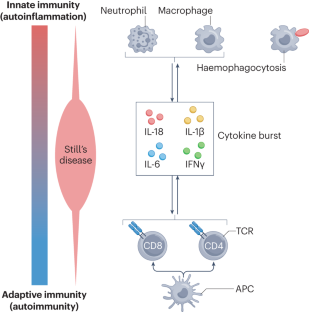
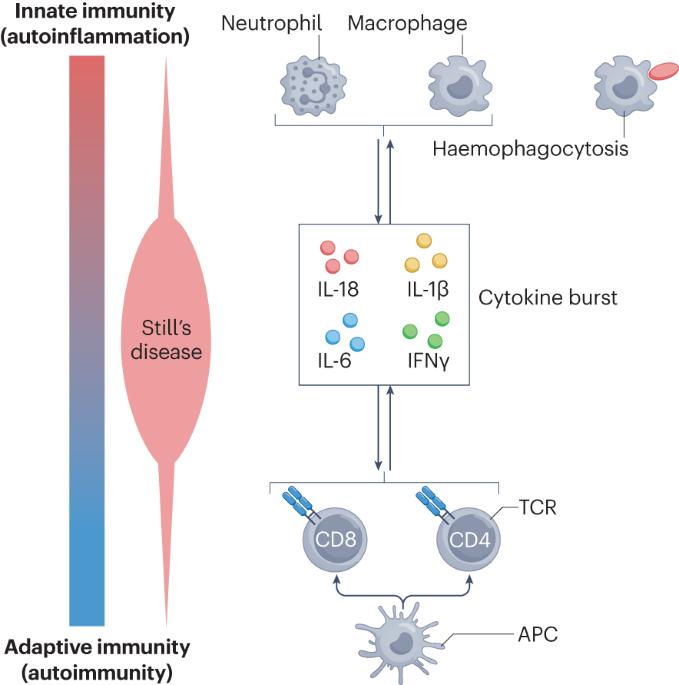
Still’s disease is a rare inflammatory syndrome that encompasses systemic juvenile idiopathic arthritis and adult-onset Still’s disease, both of which can exhibit life-threatening complications, including macrophage activation syndrome (MAS), a secondary form of haemophagocytic lymphohistiocytosis. Genetic insights into Still’s disease involve both HLA and non-HLA susceptibility genes, suggesting the involvement of adaptive immune cell-mediated immunity. At the same time, phenotypic evidence indicates the involvement of autoinflammatory processes. Evidence also implicates the type I interferon signature, mechanistic target of rapamycin complex 1 signalling and ferritin in the pathogenesis of Still’s disease and MAS. Pathological entities associated with Still’s disease include lung disease that could be associated with biologic DMARDs and with the occurrence of MAS. Historically, monophasic, recurrent and persistent Still’s disease courses were recognized. Newer proposals of alternative Still’s disease clusters could enable better dissection of clinical heterogeneity on the basis of immune cell profiles that could represent diverse endotypes or phases of disease activity. Therapeutically, data on IL-1 and IL-6 antagonism and Janus kinase inhibition suggest the importance of early administration in Still’s disease. Furthermore, there is evidence that patients who develop MAS can be treated with IFNγ antagonism. Despite these developments, unmet needs remain that can form the basis for the design of future studies leading to improvement of disease management. In this Review, the authors describe shared pathophysiology of systemic juvenile idiopathic arthritis and adult-onset Still’s disease and their life-threatening complication, macrophage activation syndrome. Therapeutic developments now enable the targeting of multiple pathways in these conditions, and evidence suggests that early use of DMARDs has the potential to prevent chronic disease.
期刊介绍:
Nature Reviews Rheumatology is part of the Nature Reviews portfolio of journals. The journal scope covers the entire spectrum of rheumatology research. We ensure that our articles are accessible to the widest possible audience.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
