IF 2.6 4区 生物学Q4 BIOCHEMISTRY & MOLECULAR BIOLOGYYeastPub Date : 2024-04-01Epub Date: 2024-01-28DOI:10.1002/yea.3927
Leandra Brettner, Rachel Eder, Kara Schmidlin, Kerry Geiler-Samerotte
{"title":"酵母中的超高通量、大规模多路复用、单细胞 RNA-seq 平台。","authors":"Leandra Brettner, Rachel Eder, Kara Schmidlin, Kerry Geiler-Samerotte","doi":"10.1002/yea.3927","DOIUrl":null,"url":null,"abstract":"<p><p>Yeasts are naturally diverse, genetically tractable, and easy to grow such that researchers can investigate any number of genotypes, environments, or interactions thereof. However, studies of yeast transcriptomes have been limited by the processing capabilities of traditional RNA sequencing techniques. Here we optimize a powerful, high-throughput single-cell RNA sequencing (scRNAseq) platform, SPLiT-seq (Split Pool Ligation-based Transcriptome sequencing), for yeasts and apply it to 43,388 cells of multiple species and ploidies. This platform utilizes a combinatorial barcoding strategy to enable massively parallel RNA sequencing of hundreds of yeast genotypes or growth conditions at once. This method can be applied to most species or strains of yeast for a fraction of the cost of traditional scRNAseq approaches. Thus, our technology permits researchers to leverage \"the awesome power of yeast\" by allowing us to survey the transcriptome of hundreds of strains and environments in a short period of time and with no specialized equipment. The key to this method is that sequential barcodes are probabilistically appended to cDNA copies of RNA while the molecules remain trapped inside of each cell. Thus, the transcriptome of each cell is labeled with a unique combination of barcodes. Since SPLiT-seq uses the cell membrane as a container for this reaction, many cells can be processed together without the need to physically isolate them from one another in separate wells or droplets. Further, the first barcode in the sequence can be chosen intentionally to identify samples from different environments or genetic backgrounds, enabling multiplexing of hundreds of unique perturbations in a single experiment. In addition to greater multiplexing capabilities, our method also facilitates a deeper investigation of biological heterogeneity, given its single-cell nature. For example, in the data presented here, we detect transcriptionally distinct cell states related to cell cycle, ploidy, metabolic strategies, and so forth, all within clonal yeast populations grown in the same environment. Hence, our technology has two obvious and impactful applications for yeast research: the first is the general study of transcriptional phenotypes across many strains and environments, and the second is investigating cell-to-cell heterogeneity across the entire transcriptome.</p>","PeriodicalId":23870,"journal":{"name":"Yeast","volume":" ","pages":"242-255"},"PeriodicalIF":2.6000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11146634/pdf/","citationCount":"0","resultStr":"{\"title\":\"An ultra high-throughput, massively multiplexable, single-cell RNA-seq platform in yeasts.\",\"authors\":\"Leandra Brettner, Rachel Eder, Kara Schmidlin, Kerry Geiler-Samerotte\",\"doi\":\"10.1002/yea.3927\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Yeasts are naturally diverse, genetically tractable, and easy to grow such that researchers can investigate any number of genotypes, environments, or interactions thereof. However, studies of yeast transcriptomes have been limited by the processing capabilities of traditional RNA sequencing techniques. Here we optimize a powerful, high-throughput single-cell RNA sequencing (scRNAseq) platform, SPLiT-seq (Split Pool Ligation-based Transcriptome sequencing), for yeasts and apply it to 43,388 cells of multiple species and ploidies. This platform utilizes a combinatorial barcoding strategy to enable massively parallel RNA sequencing of hundreds of yeast genotypes or growth conditions at once. This method can be applied to most species or strains of yeast for a fraction of the cost of traditional scRNAseq approaches. Thus, our technology permits researchers to leverage \\\"the awesome power of yeast\\\" by allowing us to survey the transcriptome of hundreds of strains and environments in a short period of time and with no specialized equipment. The key to this method is that sequential barcodes are probabilistically appended to cDNA copies of RNA while the molecules remain trapped inside of each cell. Thus, the transcriptome of each cell is labeled with a unique combination of barcodes. Since SPLiT-seq uses the cell membrane as a container for this reaction, many cells can be processed together without the need to physically isolate them from one another in separate wells or droplets. Further, the first barcode in the sequence can be chosen intentionally to identify samples from different environments or genetic backgrounds, enabling multiplexing of hundreds of unique perturbations in a single experiment. In addition to greater multiplexing capabilities, our method also facilitates a deeper investigation of biological heterogeneity, given its single-cell nature. For example, in the data presented here, we detect transcriptionally distinct cell states related to cell cycle, ploidy, metabolic strategies, and so forth, all within clonal yeast populations grown in the same environment. Hence, our technology has two obvious and impactful applications for yeast research: the first is the general study of transcriptional phenotypes across many strains and environments, and the second is investigating cell-to-cell heterogeneity across the entire transcriptome.</p>\",\"PeriodicalId\":23870,\"journal\":{\"name\":\"Yeast\",\"volume\":\" \",\"pages\":\"242-255\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11146634/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Yeast\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1002/yea.3927\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Yeast","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1002/yea.3927","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/28 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
An ultra high-throughput, massively multiplexable, single-cell RNA-seq platform in yeasts.
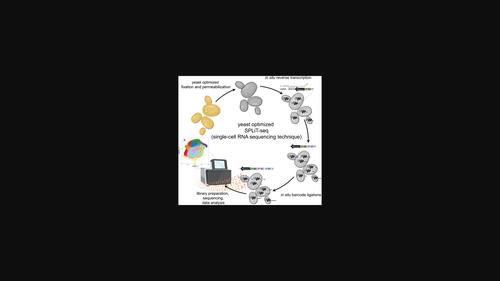
Yeasts are naturally diverse, genetically tractable, and easy to grow such that researchers can investigate any number of genotypes, environments, or interactions thereof. However, studies of yeast transcriptomes have been limited by the processing capabilities of traditional RNA sequencing techniques. Here we optimize a powerful, high-throughput single-cell RNA sequencing (scRNAseq) platform, SPLiT-seq (Split Pool Ligation-based Transcriptome sequencing), for yeasts and apply it to 43,388 cells of multiple species and ploidies. This platform utilizes a combinatorial barcoding strategy to enable massively parallel RNA sequencing of hundreds of yeast genotypes or growth conditions at once. This method can be applied to most species or strains of yeast for a fraction of the cost of traditional scRNAseq approaches. Thus, our technology permits researchers to leverage "the awesome power of yeast" by allowing us to survey the transcriptome of hundreds of strains and environments in a short period of time and with no specialized equipment. The key to this method is that sequential barcodes are probabilistically appended to cDNA copies of RNA while the molecules remain trapped inside of each cell. Thus, the transcriptome of each cell is labeled with a unique combination of barcodes. Since SPLiT-seq uses the cell membrane as a container for this reaction, many cells can be processed together without the need to physically isolate them from one another in separate wells or droplets. Further, the first barcode in the sequence can be chosen intentionally to identify samples from different environments or genetic backgrounds, enabling multiplexing of hundreds of unique perturbations in a single experiment. In addition to greater multiplexing capabilities, our method also facilitates a deeper investigation of biological heterogeneity, given its single-cell nature. For example, in the data presented here, we detect transcriptionally distinct cell states related to cell cycle, ploidy, metabolic strategies, and so forth, all within clonal yeast populations grown in the same environment. Hence, our technology has two obvious and impactful applications for yeast research: the first is the general study of transcriptional phenotypes across many strains and environments, and the second is investigating cell-to-cell heterogeneity across the entire transcriptome.
期刊介绍:
Yeast publishes original articles and reviews on the most significant developments of research with unicellular fungi, including innovative methods of broad applicability. It is essential reading for those wishing to keep up to date with this rapidly moving field of yeast biology.
Topics covered include: biochemistry and molecular biology; biodiversity and taxonomy; biotechnology; cell and developmental biology; ecology and evolution; genetics and genomics; metabolism and physiology; pathobiology; synthetic and systems biology; tools and resources




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
