{"title":"瞬时受体电位类香草素1抑制剂可抑制脑出血后神经元凋亡,从而减轻脑损伤。","authors":"Chien-Cheng Chen, Chia-Hua Ke, Chun-Hu Wu, Hung-Fu Lee, Yuan Chao, Min-Chien Tsai, Song-Kun Shyue, Szu-Fu Chen","doi":"10.1111/bpa.13244","DOIUrl":null,"url":null,"abstract":"<p>Intracerebral hemorrhage (ICH) induces a complex sequence of apoptotic cascades and inflammatory responses, leading to neurological impairment. Transient receptor potential vanilloid 1 (TRPV1), a nonselective cation channel with high calcium permeability, has been implicated in neuronal apoptosis and inflammatory responses. This study used a mouse ICH model and neuronal cultures to examine whether TRPV1 activation exacerbates brain damage and neurological deficits by promoting neuronal apoptosis and neuroinflammation. ICH was induced by injecting collagenase in both wild-type (WT) C57BL/6 mice and TRPV1<sup>−/−</sup> mice. Capsaicin (CAP; a TRPV1 agonist) or capsazepine (a TRPV1 antagonist) was administered by intracerebroventricular injection 30 min before ICH induction in WT mice. The effects of genetic deletion or pharmacological inhibition of TRPV1 using CAP or capsazepine on motor deficits, histological damage, apoptotic responses, blood–brain barrier (BBB) permeability, and neuroinflammatory reactions were explored. The antiapoptotic mechanisms and calcium influx induced by TRPV1 inactivation were investigated in cultured hemin-stimulated neurons. TRPV1 expression was upregulated in the hemorrhagic brain, and TRPV1 was expressed in neurons, microglia, and astrocytes after ICH. Genetic deletion of TRPV1 significantly attenuated motor deficits and brain atrophy for up to 28 days. Deletion of TRPV1 also reduced brain damage, neurodegeneration, microglial activation, cytokine expression, and cell apoptosis at 1 day post-ICH. Similarly, the administration of CAP ameliorated brain damage, neurodegeneration, brain edema, BBB permeability, and cytokine expression at 1 day post-ICH. In primary neuronal cultures, pharmacological inactivation of TRPV1 by CAP attenuated neuronal vulnerability to hemin-induced injury, suppressed apoptosis, and preserved mitochondrial integrity in vitro. Mechanistically, CAP reduced hemin-stimulated calcium influx and prevented the phosphorylation of CaMKII in cultured neurons, which was associated with reduced activation of P38 and c-Jun NH<sub>2</sub>-terminal kinase mitogen-activated protein kinase signaling. Our results suggest that TRPV1 inhibition may be a potential therapy for ICH by suppressing mitochondria-related neuronal apoptosis.</p>","PeriodicalId":9290,"journal":{"name":"Brain Pathology","volume":"34 5","pages":""},"PeriodicalIF":5.0000,"publicationDate":"2024-02-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bpa.13244","citationCount":"0","resultStr":"{\"title\":\"Transient receptor potential vanilloid 1 inhibition reduces brain damage by suppressing neuronal apoptosis after intracerebral hemorrhage\",\"authors\":\"Chien-Cheng Chen, Chia-Hua Ke, Chun-Hu Wu, Hung-Fu Lee, Yuan Chao, Min-Chien Tsai, Song-Kun Shyue, Szu-Fu Chen\",\"doi\":\"10.1111/bpa.13244\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Intracerebral hemorrhage (ICH) induces a complex sequence of apoptotic cascades and inflammatory responses, leading to neurological impairment. Transient receptor potential vanilloid 1 (TRPV1), a nonselective cation channel with high calcium permeability, has been implicated in neuronal apoptosis and inflammatory responses. This study used a mouse ICH model and neuronal cultures to examine whether TRPV1 activation exacerbates brain damage and neurological deficits by promoting neuronal apoptosis and neuroinflammation. ICH was induced by injecting collagenase in both wild-type (WT) C57BL/6 mice and TRPV1<sup>−/−</sup> mice. Capsaicin (CAP; a TRPV1 agonist) or capsazepine (a TRPV1 antagonist) was administered by intracerebroventricular injection 30 min before ICH induction in WT mice. The effects of genetic deletion or pharmacological inhibition of TRPV1 using CAP or capsazepine on motor deficits, histological damage, apoptotic responses, blood–brain barrier (BBB) permeability, and neuroinflammatory reactions were explored. The antiapoptotic mechanisms and calcium influx induced by TRPV1 inactivation were investigated in cultured hemin-stimulated neurons. TRPV1 expression was upregulated in the hemorrhagic brain, and TRPV1 was expressed in neurons, microglia, and astrocytes after ICH. Genetic deletion of TRPV1 significantly attenuated motor deficits and brain atrophy for up to 28 days. Deletion of TRPV1 also reduced brain damage, neurodegeneration, microglial activation, cytokine expression, and cell apoptosis at 1 day post-ICH. Similarly, the administration of CAP ameliorated brain damage, neurodegeneration, brain edema, BBB permeability, and cytokine expression at 1 day post-ICH. In primary neuronal cultures, pharmacological inactivation of TRPV1 by CAP attenuated neuronal vulnerability to hemin-induced injury, suppressed apoptosis, and preserved mitochondrial integrity in vitro. Mechanistically, CAP reduced hemin-stimulated calcium influx and prevented the phosphorylation of CaMKII in cultured neurons, which was associated with reduced activation of P38 and c-Jun NH<sub>2</sub>-terminal kinase mitogen-activated protein kinase signaling. Our results suggest that TRPV1 inhibition may be a potential therapy for ICH by suppressing mitochondria-related neuronal apoptosis.</p>\",\"PeriodicalId\":9290,\"journal\":{\"name\":\"Brain Pathology\",\"volume\":\"34 5\",\"pages\":\"\"},\"PeriodicalIF\":5.0000,\"publicationDate\":\"2024-02-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bpa.13244\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Brain Pathology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/bpa.13244\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain Pathology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/bpa.13244","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
摘要
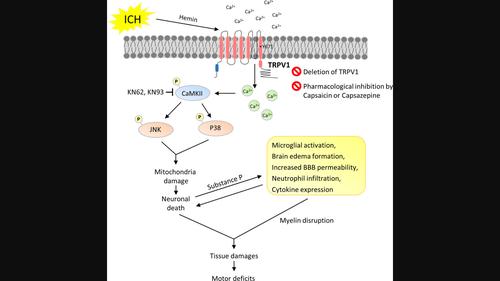
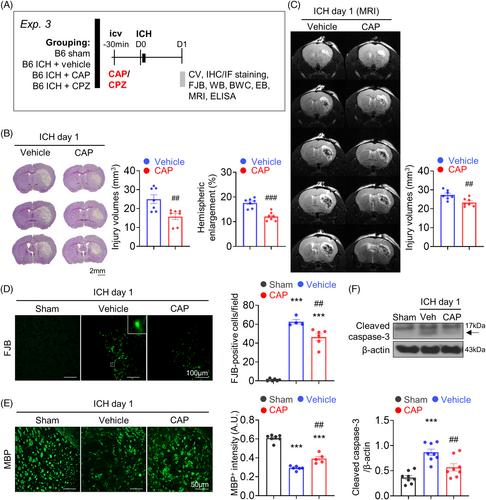
脑出血(ICH)会诱发一系列复杂的细胞凋亡级联反应和炎症反应,从而导致神经功能损伤。瞬时受体电位类香草素 1(TRPV1)是一种具有高钙通透性的非选择性阳离子通道,与神经元凋亡和炎症反应有关。本研究利用小鼠 ICH 模型和神经元培养物来研究 TRPV1 激活是否会通过促进神经元凋亡和神经炎症而加重脑损伤和神经功能缺损。通过向野生型(WT)C57BL/6小鼠和TRPV1-/-小鼠注射胶原酶诱导ICH。在诱导 WT 小鼠 ICH 前 30 分钟,通过脑室内注射辣椒素(CAP,一种 TRPV1 激动剂)或辣椒氮平(一种 TRPV1 拮抗剂)。研究人员探讨了基因缺失或使用 CAP 或卡氮平药物抑制 TRPV1 对运动障碍、组织学损伤、细胞凋亡反应、血脑屏障(BBB)通透性和神经炎症反应的影响。在培养的海明刺激神经元中,研究了 TRPV1 失活诱导的抗凋亡机制和钙离子流入。TRPV1在出血脑中表达上调,TRPV1在ICH后的神经元、小胶质细胞和星形胶质细胞中均有表达。基因缺失 TRPV1 可在长达 28 天的时间内显著减轻运动障碍和脑萎缩。基因缺失 TRPV1 还可减少 ICH 后 1 天的脑损伤、神经变性、小胶质细胞活化、细胞因子表达和细胞凋亡。同样,在脑缺血后 1 天,服用 CAP 可改善脑损伤、神经变性、脑水肿、BBB 通透性和细胞因子表达。在原代神经元培养中,CAP通过药理作用使TRPV1失活,可减轻神经元对血清素诱导的损伤的脆弱性,抑制细胞凋亡,并保护体外线粒体的完整性。从机理上讲,CAP 可减少海明刺激的钙离子流入,阻止培养神经元中 CaMKII 的磷酸化,这与 P38 和 c-Jun NH2 -terminal 激酶丝裂原活化蛋白激酶信号的激活减少有关。我们的研究结果表明,抑制 TRPV1 可抑制线粒体相关的神经元凋亡,从而成为治疗 ICH 的一种潜在疗法。
Transient receptor potential vanilloid 1 inhibition reduces brain damage by suppressing neuronal apoptosis after intracerebral hemorrhage
Intracerebral hemorrhage (ICH) induces a complex sequence of apoptotic cascades and inflammatory responses, leading to neurological impairment. Transient receptor potential vanilloid 1 (TRPV1), a nonselective cation channel with high calcium permeability, has been implicated in neuronal apoptosis and inflammatory responses. This study used a mouse ICH model and neuronal cultures to examine whether TRPV1 activation exacerbates brain damage and neurological deficits by promoting neuronal apoptosis and neuroinflammation. ICH was induced by injecting collagenase in both wild-type (WT) C57BL/6 mice and TRPV1−/− mice. Capsaicin (CAP; a TRPV1 agonist) or capsazepine (a TRPV1 antagonist) was administered by intracerebroventricular injection 30 min before ICH induction in WT mice. The effects of genetic deletion or pharmacological inhibition of TRPV1 using CAP or capsazepine on motor deficits, histological damage, apoptotic responses, blood–brain barrier (BBB) permeability, and neuroinflammatory reactions were explored. The antiapoptotic mechanisms and calcium influx induced by TRPV1 inactivation were investigated in cultured hemin-stimulated neurons. TRPV1 expression was upregulated in the hemorrhagic brain, and TRPV1 was expressed in neurons, microglia, and astrocytes after ICH. Genetic deletion of TRPV1 significantly attenuated motor deficits and brain atrophy for up to 28 days. Deletion of TRPV1 also reduced brain damage, neurodegeneration, microglial activation, cytokine expression, and cell apoptosis at 1 day post-ICH. Similarly, the administration of CAP ameliorated brain damage, neurodegeneration, brain edema, BBB permeability, and cytokine expression at 1 day post-ICH. In primary neuronal cultures, pharmacological inactivation of TRPV1 by CAP attenuated neuronal vulnerability to hemin-induced injury, suppressed apoptosis, and preserved mitochondrial integrity in vitro. Mechanistically, CAP reduced hemin-stimulated calcium influx and prevented the phosphorylation of CaMKII in cultured neurons, which was associated with reduced activation of P38 and c-Jun NH2-terminal kinase mitogen-activated protein kinase signaling. Our results suggest that TRPV1 inhibition may be a potential therapy for ICH by suppressing mitochondria-related neuronal apoptosis.
期刊介绍:
Brain Pathology is the journal of choice for biomedical scientists investigating diseases of the nervous system. The official journal of the International Society of Neuropathology, Brain Pathology is a peer-reviewed quarterly publication that includes original research, review articles and symposia focuses on the pathogenesis of neurological disease.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
