Harish Gopalakrishna, Bilal Asif, Anjali Rai, Hari S Conjeevaram, Maria Mironova, David E Kleiner, Alexandra F Freeman, Theo Heller
{"title":"Prolidase 缺乏症患者的慢性肝病:病例系列","authors":"Harish Gopalakrishna, Bilal Asif, Anjali Rai, Hari S Conjeevaram, Maria Mironova, David E Kleiner, Alexandra F Freeman, Theo Heller","doi":"10.1159/000536117","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Prolidase deficiency is a rare autosomal recessive disorder caused by variants in the <i>PEPD</i> gene. Patients usually have multi-organ involvement and a wide range of clinical features including recurrent skin ulcers, dysmorphic facial features, recurrent infections, intellectual disability, and splenomegaly. Studies have shown that patients with prolidase deficiency may have hepatic manifestations including hepatomegaly and abnormal liver enzymes. However, there is no detailed description of liver disease in this patient population.</p><p><strong>Case presentation: </strong>Here, we present 3 patients with prolidase deficiency with varying extents of hepatic involvement.</p><p><strong>Conclusion: </strong>Prolidase deficiency patients with liver disease should be followed up long term to understand more about the pathophysiology and the impact of liver disease on long-term outcomes.</p>","PeriodicalId":9614,"journal":{"name":"Case Reports in Gastroenterology","volume":"18 1","pages":"49-57"},"PeriodicalIF":0.7000,"publicationDate":"2024-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10834036/pdf/","citationCount":"0","resultStr":"{\"title\":\"Chronic Liver Disease in Patients with Prolidase Deficiency: A Case Series.\",\"authors\":\"Harish Gopalakrishna, Bilal Asif, Anjali Rai, Hari S Conjeevaram, Maria Mironova, David E Kleiner, Alexandra F Freeman, Theo Heller\",\"doi\":\"10.1159/000536117\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>Prolidase deficiency is a rare autosomal recessive disorder caused by variants in the <i>PEPD</i> gene. Patients usually have multi-organ involvement and a wide range of clinical features including recurrent skin ulcers, dysmorphic facial features, recurrent infections, intellectual disability, and splenomegaly. Studies have shown that patients with prolidase deficiency may have hepatic manifestations including hepatomegaly and abnormal liver enzymes. However, there is no detailed description of liver disease in this patient population.</p><p><strong>Case presentation: </strong>Here, we present 3 patients with prolidase deficiency with varying extents of hepatic involvement.</p><p><strong>Conclusion: </strong>Prolidase deficiency patients with liver disease should be followed up long term to understand more about the pathophysiology and the impact of liver disease on long-term outcomes.</p>\",\"PeriodicalId\":9614,\"journal\":{\"name\":\"Case Reports in Gastroenterology\",\"volume\":\"18 1\",\"pages\":\"49-57\"},\"PeriodicalIF\":0.7000,\"publicationDate\":\"2024-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10834036/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Gastroenterology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1159/000536117\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"GASTROENTEROLOGY & HEPATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Gastroenterology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000536117","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GASTROENTEROLOGY & HEPATOLOGY","Score":null,"Total":0}
Chronic Liver Disease in Patients with Prolidase Deficiency: A Case Series.
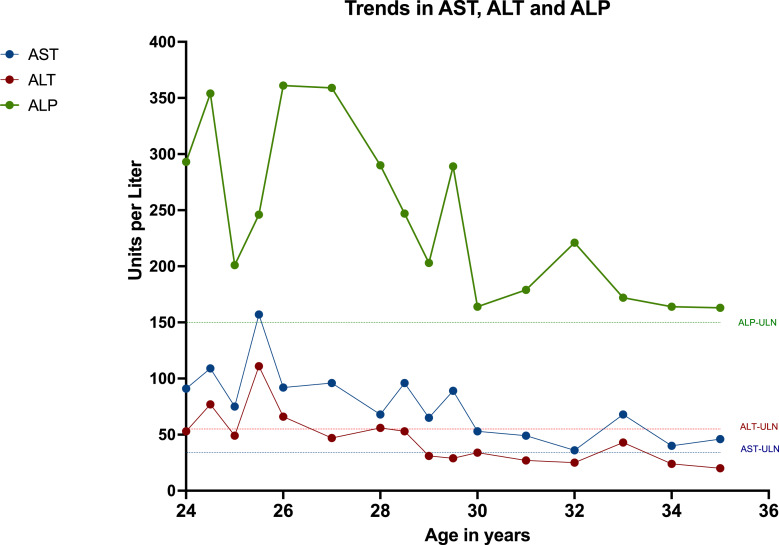
Introduction: Prolidase deficiency is a rare autosomal recessive disorder caused by variants in the PEPD gene. Patients usually have multi-organ involvement and a wide range of clinical features including recurrent skin ulcers, dysmorphic facial features, recurrent infections, intellectual disability, and splenomegaly. Studies have shown that patients with prolidase deficiency may have hepatic manifestations including hepatomegaly and abnormal liver enzymes. However, there is no detailed description of liver disease in this patient population.
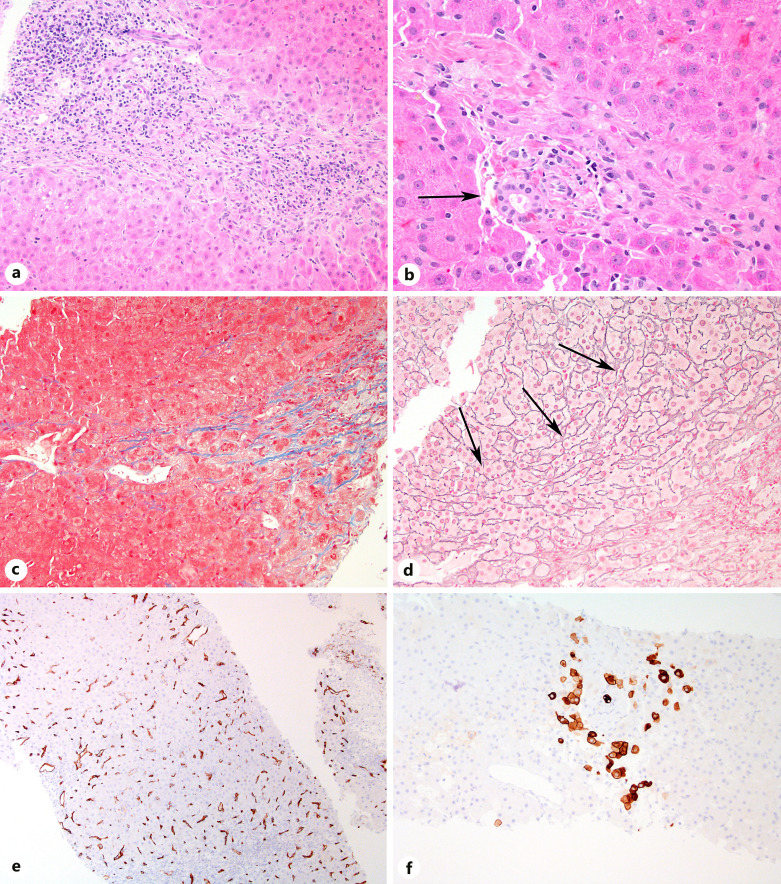
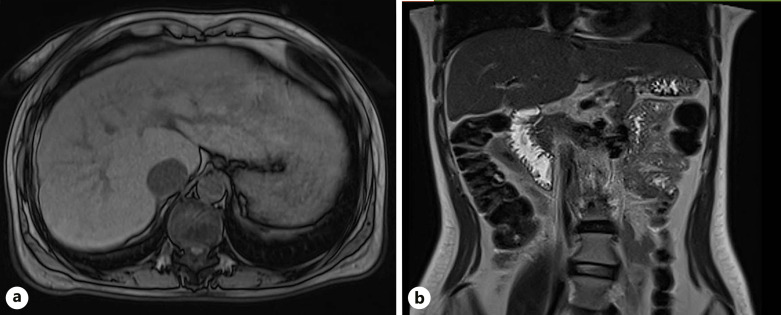
Case presentation: Here, we present 3 patients with prolidase deficiency with varying extents of hepatic involvement.
Conclusion: Prolidase deficiency patients with liver disease should be followed up long term to understand more about the pathophysiology and the impact of liver disease on long-term outcomes.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
