Artemis Efstratiou, Arnaud Gaigher, Sven Künzel, Ana Teles, Tobias L. Lenz
{"title":"对 NGS 基因分型管道进行模板特异性优化,揭示了 MHC 基因表达的等位基因特异性变异。","authors":"Artemis Efstratiou, Arnaud Gaigher, Sven Künzel, Ana Teles, Tobias L. Lenz","doi":"10.1111/1755-0998.13935","DOIUrl":null,"url":null,"abstract":"<p>Using high-throughput sequencing for precise genotyping of multi-locus gene families, such as the major histocompatibility complex (MHC), remains challenging, due to the complexity of the data and difficulties in distinguishing genuine from erroneous variants. Several dedicated genotyping pipelines for data from high-throughput sequencing, such as next-generation sequencing (NGS), have been developed to tackle the ensuing risk of artificially inflated diversity. Here, we thoroughly assess three such multi-locus genotyping pipelines for NGS data, the DOC method, AmpliSAS and ACACIA, using MHC class IIβ data sets of three-spined stickleback gDNA, cDNA and “artificial” plasmid samples with known allelic diversity. We show that genotyping of gDNA and plasmid samples at optimal pipeline parameters was highly accurate and reproducible across methods. However, for cDNA data, the gDNA-optimal parameter configuration yielded decreased overall genotyping precision and consistency between pipelines. Further adjustments of key clustering parameters were required tο account for higher error rates and larger variation in sequencing depth per allele, highlighting the importance of template-specific pipeline optimization for reliable genotyping of multi-locus gene families. Through accurate paired gDNA-cDNA typing and MHC-II haplotype inference, we show that MHC-II allele-specific expression levels correlate negatively with allele number across haplotypes. Lastly, sibship-assisted cDNA-typing of MHC-I revealed novel variants linked in haplotype blocks, and a higher-than-previously-reported individual MHC-I allelic diversity. In conclusion, we provide novel genotyping protocols for the three-spined stickleback MHC-I and -II genes, and evaluate the performance of popular NGS-genotyping pipelines. We also show that fine-tuned genotyping of paired gDNA-cDNA samples facilitates amplification bias-corrected MHC allele expression analysis.</p>","PeriodicalId":211,"journal":{"name":"Molecular Ecology Resources","volume":"24 4","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-02-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.13935","citationCount":"0","resultStr":"{\"title\":\"Template-specific optimization of NGS genotyping pipelines reveals allele-specific variation in MHC gene expression\",\"authors\":\"Artemis Efstratiou, Arnaud Gaigher, Sven Künzel, Ana Teles, Tobias L. Lenz\",\"doi\":\"10.1111/1755-0998.13935\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Using high-throughput sequencing for precise genotyping of multi-locus gene families, such as the major histocompatibility complex (MHC), remains challenging, due to the complexity of the data and difficulties in distinguishing genuine from erroneous variants. Several dedicated genotyping pipelines for data from high-throughput sequencing, such as next-generation sequencing (NGS), have been developed to tackle the ensuing risk of artificially inflated diversity. Here, we thoroughly assess three such multi-locus genotyping pipelines for NGS data, the DOC method, AmpliSAS and ACACIA, using MHC class IIβ data sets of three-spined stickleback gDNA, cDNA and “artificial” plasmid samples with known allelic diversity. We show that genotyping of gDNA and plasmid samples at optimal pipeline parameters was highly accurate and reproducible across methods. However, for cDNA data, the gDNA-optimal parameter configuration yielded decreased overall genotyping precision and consistency between pipelines. Further adjustments of key clustering parameters were required tο account for higher error rates and larger variation in sequencing depth per allele, highlighting the importance of template-specific pipeline optimization for reliable genotyping of multi-locus gene families. Through accurate paired gDNA-cDNA typing and MHC-II haplotype inference, we show that MHC-II allele-specific expression levels correlate negatively with allele number across haplotypes. Lastly, sibship-assisted cDNA-typing of MHC-I revealed novel variants linked in haplotype blocks, and a higher-than-previously-reported individual MHC-I allelic diversity. In conclusion, we provide novel genotyping protocols for the three-spined stickleback MHC-I and -II genes, and evaluate the performance of popular NGS-genotyping pipelines. We also show that fine-tuned genotyping of paired gDNA-cDNA samples facilitates amplification bias-corrected MHC allele expression analysis.</p>\",\"PeriodicalId\":211,\"journal\":{\"name\":\"Molecular Ecology Resources\",\"volume\":\"24 4\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-02-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.13935\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Ecology Resources\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13935\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Ecology Resources","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13935","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Template-specific optimization of NGS genotyping pipelines reveals allele-specific variation in MHC gene expression
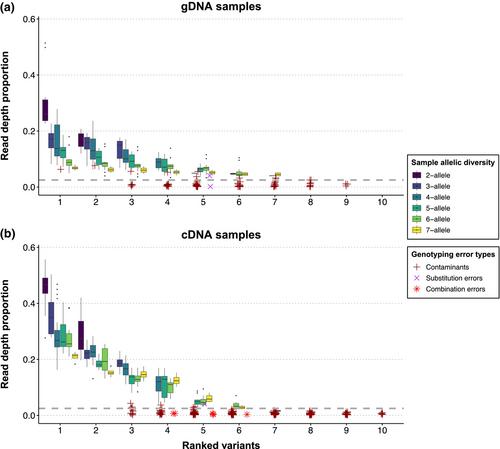
Using high-throughput sequencing for precise genotyping of multi-locus gene families, such as the major histocompatibility complex (MHC), remains challenging, due to the complexity of the data and difficulties in distinguishing genuine from erroneous variants. Several dedicated genotyping pipelines for data from high-throughput sequencing, such as next-generation sequencing (NGS), have been developed to tackle the ensuing risk of artificially inflated diversity. Here, we thoroughly assess three such multi-locus genotyping pipelines for NGS data, the DOC method, AmpliSAS and ACACIA, using MHC class IIβ data sets of three-spined stickleback gDNA, cDNA and “artificial” plasmid samples with known allelic diversity. We show that genotyping of gDNA and plasmid samples at optimal pipeline parameters was highly accurate and reproducible across methods. However, for cDNA data, the gDNA-optimal parameter configuration yielded decreased overall genotyping precision and consistency between pipelines. Further adjustments of key clustering parameters were required tο account for higher error rates and larger variation in sequencing depth per allele, highlighting the importance of template-specific pipeline optimization for reliable genotyping of multi-locus gene families. Through accurate paired gDNA-cDNA typing and MHC-II haplotype inference, we show that MHC-II allele-specific expression levels correlate negatively with allele number across haplotypes. Lastly, sibship-assisted cDNA-typing of MHC-I revealed novel variants linked in haplotype blocks, and a higher-than-previously-reported individual MHC-I allelic diversity. In conclusion, we provide novel genotyping protocols for the three-spined stickleback MHC-I and -II genes, and evaluate the performance of popular NGS-genotyping pipelines. We also show that fine-tuned genotyping of paired gDNA-cDNA samples facilitates amplification bias-corrected MHC allele expression analysis.
期刊介绍:
Molecular Ecology Resources promotes the creation of comprehensive resources for the scientific community, encompassing computer programs, statistical and molecular advancements, and a diverse array of molecular tools. Serving as a conduit for disseminating these resources, the journal targets a broad audience of researchers in the fields of evolution, ecology, and conservation. Articles in Molecular Ecology Resources are crafted to support investigations tackling significant questions within these disciplines.
In addition to original resource articles, Molecular Ecology Resources features Reviews, Opinions, and Comments relevant to the field. The journal also periodically releases Special Issues focusing on resource development within specific areas.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
