{"title":"遗传性痉挛性截瘫 46 型 (SPG46):意大利大型病例系列中的新 GBA2 变体及文献综述。","authors":"Ettore Cioffi, Gianluca Coppola, Olimpia Musumeci, Salvatore Gallone, Gabriella Silvestri, Salvatore Rossi, Fiorella Piemonte, Jessica D'Amico, Alessandra Tessa, Filippo Maria Santorelli, Carlo Casali","doi":"10.1007/s10048-024-00749-9","DOIUrl":null,"url":null,"abstract":"<p><p>Hereditary spastic paraparesis (HSP) is a group of central nervous system diseases primarily affecting the spinal upper motor neurons, with different inheritance patterns and phenotypes. SPG46 is a rare, early-onset and autosomal recessive HSP, linked to biallelic GBA2 mutations. About thirty families have been described worldwide, with different phenotypes like complicated HSP, recessive cerebellar ataxia or Marinesco-Sjögren Syndrome. Herein, we report five SPG46 patients harbouring five novel GBA2 mutations, the largest series described in Italy so far. Probands were enrolled in five different centres and underwent neurological examination, clinical cognitive assessment, column imaging for scoliosis assessment, ophthalmologic examination, brain imaging, GBA2 activity in peripheral blood cells and genetic testing. Their phenotype was consistent with HSP, with notable features like upper gaze palsy and movement disorders. We review demographic, genetic, biochemical and clinical information from all documented cases in the existing literature, focusing on the global distribution of cases, the features of the syndrome, its variable presentation, new potential identifying features and the significance of measuring GBA2 enzyme activity.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"51-67"},"PeriodicalIF":1.2000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11076336/pdf/","citationCount":"0","resultStr":"{\"title\":\"Hereditary spastic paraparesis type 46 (SPG46): new GBA2 variants in a large Italian case series and review of the literature.\",\"authors\":\"Ettore Cioffi, Gianluca Coppola, Olimpia Musumeci, Salvatore Gallone, Gabriella Silvestri, Salvatore Rossi, Fiorella Piemonte, Jessica D'Amico, Alessandra Tessa, Filippo Maria Santorelli, Carlo Casali\",\"doi\":\"10.1007/s10048-024-00749-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hereditary spastic paraparesis (HSP) is a group of central nervous system diseases primarily affecting the spinal upper motor neurons, with different inheritance patterns and phenotypes. SPG46 is a rare, early-onset and autosomal recessive HSP, linked to biallelic GBA2 mutations. About thirty families have been described worldwide, with different phenotypes like complicated HSP, recessive cerebellar ataxia or Marinesco-Sjögren Syndrome. Herein, we report five SPG46 patients harbouring five novel GBA2 mutations, the largest series described in Italy so far. Probands were enrolled in five different centres and underwent neurological examination, clinical cognitive assessment, column imaging for scoliosis assessment, ophthalmologic examination, brain imaging, GBA2 activity in peripheral blood cells and genetic testing. Their phenotype was consistent with HSP, with notable features like upper gaze palsy and movement disorders. We review demographic, genetic, biochemical and clinical information from all documented cases in the existing literature, focusing on the global distribution of cases, the features of the syndrome, its variable presentation, new potential identifying features and the significance of measuring GBA2 enzyme activity.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\" \",\"pages\":\"51-67\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11076336/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-024-00749-9\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/2/9 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-024-00749-9","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/2/9 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Hereditary spastic paraparesis type 46 (SPG46): new GBA2 variants in a large Italian case series and review of the literature.
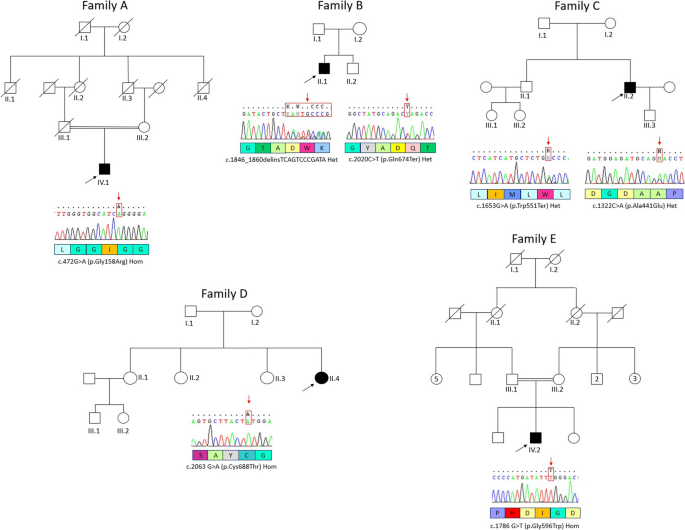
Hereditary spastic paraparesis (HSP) is a group of central nervous system diseases primarily affecting the spinal upper motor neurons, with different inheritance patterns and phenotypes. SPG46 is a rare, early-onset and autosomal recessive HSP, linked to biallelic GBA2 mutations. About thirty families have been described worldwide, with different phenotypes like complicated HSP, recessive cerebellar ataxia or Marinesco-Sjögren Syndrome. Herein, we report five SPG46 patients harbouring five novel GBA2 mutations, the largest series described in Italy so far. Probands were enrolled in five different centres and underwent neurological examination, clinical cognitive assessment, column imaging for scoliosis assessment, ophthalmologic examination, brain imaging, GBA2 activity in peripheral blood cells and genetic testing. Their phenotype was consistent with HSP, with notable features like upper gaze palsy and movement disorders. We review demographic, genetic, biochemical and clinical information from all documented cases in the existing literature, focusing on the global distribution of cases, the features of the syndrome, its variable presentation, new potential identifying features and the significance of measuring GBA2 enzyme activity.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
