Bryan K Ward, Kirsten A Loffell, John P Walsh, Warwick D Howe, Suzanne J Brown, Scott G Wilson
{"title":"病例介绍:对在一名高钙血症患者身上发现的 CASR 变异体进行功能评估,证实该患者和一个曾被误诊为原发性甲状旁腺功能亢进症的姐妹患有家族性高钙尿酸性高钙血症。","authors":"Bryan K Ward, Kirsten A Loffell, John P Walsh, Warwick D Howe, Suzanne J Brown, Scott G Wilson","doi":"10.1155/2024/6652801","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Primary hyperparathyroidism (PHPT) and familial hypocalciuric hypercalcaemia (FHH) are common causes of hypercalcaemia. Patients are mostly asymptomatic in the case of FHH and often so in the case of PHPT. In addition, biochemical parameters show considerable overlap, making differential diagnosis difficult. Genetic screening for inactivating variants in the calcium-sensing receptor (<i>CASR</i>) gene that are causative of FHH assists with the diagnosis since such variants are not generally associated with PHPT. However, novel <i>CASR</i> variants must undergo functional assessment before they can be definitively assigned a causative role in FHH. <i>Case Presentations</i>. We describe a 73-year-old female (patient A) who presented with mild parathyroid hormone (PTH)-dependent hypercalcaemia and a history of osteoporosis. Family history revealed that her sister (patient B) had presented a decade earlier with symptoms of PHPT including a history of mild hypercalcaemia and multiple renal calculi, prompting parathyroid surgery. However, a subtotal parathyroidectomy did not resolve her hypercalcaemia long term. On this basis, genetic screening was performed on patient A. This identified a heterozygous variant in the <i>CASR</i>, NM_000388.4:c.T101C: p.Leu34Pro (L34P). Functional analysis showed that the L34P variant was unable to produce mature, dimerized receptor and did not respond to Ca<sup>++</sup> ions. Adopting American College of Medical Genetics-based guidelines, the variant was classified as 'Pathogenic (II)'. Patient B was subsequently found to carry the L34P variant heterozygously, confirming a diagnosis of FHH, not PHPT.</p><p><strong>Conclusion: </strong>This study shows the importance of examining patient's family history in providing clues to the diagnosis in isolated cases of hypercalcaemia. In this case, history of a sister's unsuccessful parathyroidectomy prompted genetic screening in a patient who might otherwise have undergone inappropriate parathyroid surgery. Screening detected an inactivating <i>CASR</i> variant, firming up a diagnosis of FHH. These studies reaffirm the requirement for functionally assessing novel <i>CASR</i> variants prior to assigning causality to FHH.</p>","PeriodicalId":9621,"journal":{"name":"Case Reports in Endocrinology","volume":"2024 ","pages":"6652801"},"PeriodicalIF":0.9000,"publicationDate":"2024-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10858793/pdf/","citationCount":"0","resultStr":"{\"title\":\"Case Presentation: Functional Assessment of a <i>CASR</i> Variant Identified in a Patient with Hypercalcaemia Confirms Familial Hypocalciuric Hypercalcaemia in the Patient and a Sister Previously Misdiagnosed with Primary Hyperparathyroidism.\",\"authors\":\"Bryan K Ward, Kirsten A Loffell, John P Walsh, Warwick D Howe, Suzanne J Brown, Scott G Wilson\",\"doi\":\"10.1155/2024/6652801\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Primary hyperparathyroidism (PHPT) and familial hypocalciuric hypercalcaemia (FHH) are common causes of hypercalcaemia. Patients are mostly asymptomatic in the case of FHH and often so in the case of PHPT. In addition, biochemical parameters show considerable overlap, making differential diagnosis difficult. Genetic screening for inactivating variants in the calcium-sensing receptor (<i>CASR</i>) gene that are causative of FHH assists with the diagnosis since such variants are not generally associated with PHPT. However, novel <i>CASR</i> variants must undergo functional assessment before they can be definitively assigned a causative role in FHH. <i>Case Presentations</i>. We describe a 73-year-old female (patient A) who presented with mild parathyroid hormone (PTH)-dependent hypercalcaemia and a history of osteoporosis. Family history revealed that her sister (patient B) had presented a decade earlier with symptoms of PHPT including a history of mild hypercalcaemia and multiple renal calculi, prompting parathyroid surgery. However, a subtotal parathyroidectomy did not resolve her hypercalcaemia long term. On this basis, genetic screening was performed on patient A. This identified a heterozygous variant in the <i>CASR</i>, NM_000388.4:c.T101C: p.Leu34Pro (L34P). Functional analysis showed that the L34P variant was unable to produce mature, dimerized receptor and did not respond to Ca<sup>++</sup> ions. Adopting American College of Medical Genetics-based guidelines, the variant was classified as 'Pathogenic (II)'. Patient B was subsequently found to carry the L34P variant heterozygously, confirming a diagnosis of FHH, not PHPT.</p><p><strong>Conclusion: </strong>This study shows the importance of examining patient's family history in providing clues to the diagnosis in isolated cases of hypercalcaemia. In this case, history of a sister's unsuccessful parathyroidectomy prompted genetic screening in a patient who might otherwise have undergone inappropriate parathyroid surgery. Screening detected an inactivating <i>CASR</i> variant, firming up a diagnosis of FHH. These studies reaffirm the requirement for functionally assessing novel <i>CASR</i> variants prior to assigning causality to FHH.</p>\",\"PeriodicalId\":9621,\"journal\":{\"name\":\"Case Reports in Endocrinology\",\"volume\":\"2024 \",\"pages\":\"6652801\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2024-02-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10858793/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2024/6652801\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2024/6652801","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
背景:原发性甲状旁腺功能亢进症(PHPT)和家族性低钙血症(FHH)是导致高钙血症的常见原因。FHH 患者大多无症状,而 PHPT 患者通常也无症状。此外,生化指标显示出相当大的重叠性,给鉴别诊断带来困难。基因筛查钙感受体(CASR)基因中导致 FHH 的失活变体有助于诊断,因为这类变体通常与 PHPT 无关。然而,新型 CASR 变体必须经过功能评估后才能明确其在 FHH 中的致病作用。病例介绍。我们描述了一名 73 岁的女性(患者 A),她患有轻度甲状旁腺激素(PTH)依赖性高钙血症和骨质疏松症。家族病史显示,她的姐姐(患者B)十年前曾出现PHPT症状,包括轻度高钙血症和多发性肾结石,并因此接受了甲状旁腺手术。然而,甲状旁腺次全切除术并没有长期缓解她的高钙血症。在此基础上,对患者A进行了基因筛查,发现了CASR的杂合变异体NM_000388.4:c.T101C: p.Leu34Pro(L34P)。功能分析显示,L34P 变体无法产生成熟的二聚化受体,对 Ca++ 离子也没有反应。根据美国医学遗传学会(American College of Medical Genetics)的指导方针,该变体被归类为 "致病性(II)"。随后发现患者 B 杂合携带 L34P 变体,确诊为 FHH,而非 PHPT:这项研究表明,在孤立的高钙血症病例中,检查患者的家族史对提供诊断线索非常重要。在本病例中,患者的姐姐曾接受过甲状旁腺切除术,但未获成功,这促使她接受了基因筛查,否则她可能会接受不适当的甲状旁腺手术。筛查发现了一个失活的CASR变异体,从而确定了FHH的诊断。这些研究再次证明,在确定 FHH 的因果关系之前,需要对新型 CASR 变体进行功能评估。
Case Presentation: Functional Assessment of a CASR Variant Identified in a Patient with Hypercalcaemia Confirms Familial Hypocalciuric Hypercalcaemia in the Patient and a Sister Previously Misdiagnosed with Primary Hyperparathyroidism.
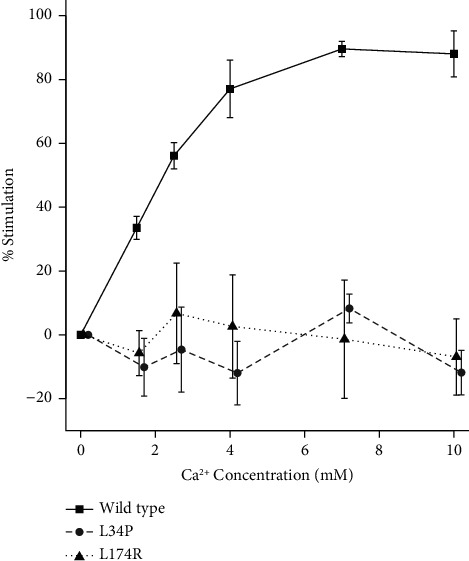
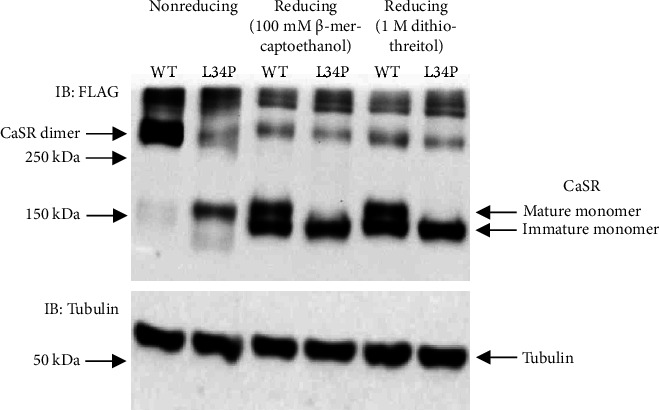
Background: Primary hyperparathyroidism (PHPT) and familial hypocalciuric hypercalcaemia (FHH) are common causes of hypercalcaemia. Patients are mostly asymptomatic in the case of FHH and often so in the case of PHPT. In addition, biochemical parameters show considerable overlap, making differential diagnosis difficult. Genetic screening for inactivating variants in the calcium-sensing receptor (CASR) gene that are causative of FHH assists with the diagnosis since such variants are not generally associated with PHPT. However, novel CASR variants must undergo functional assessment before they can be definitively assigned a causative role in FHH. Case Presentations. We describe a 73-year-old female (patient A) who presented with mild parathyroid hormone (PTH)-dependent hypercalcaemia and a history of osteoporosis. Family history revealed that her sister (patient B) had presented a decade earlier with symptoms of PHPT including a history of mild hypercalcaemia and multiple renal calculi, prompting parathyroid surgery. However, a subtotal parathyroidectomy did not resolve her hypercalcaemia long term. On this basis, genetic screening was performed on patient A. This identified a heterozygous variant in the CASR, NM_000388.4:c.T101C: p.Leu34Pro (L34P). Functional analysis showed that the L34P variant was unable to produce mature, dimerized receptor and did not respond to Ca++ ions. Adopting American College of Medical Genetics-based guidelines, the variant was classified as 'Pathogenic (II)'. Patient B was subsequently found to carry the L34P variant heterozygously, confirming a diagnosis of FHH, not PHPT.
Conclusion: This study shows the importance of examining patient's family history in providing clues to the diagnosis in isolated cases of hypercalcaemia. In this case, history of a sister's unsuccessful parathyroidectomy prompted genetic screening in a patient who might otherwise have undergone inappropriate parathyroid surgery. Screening detected an inactivating CASR variant, firming up a diagnosis of FHH. These studies reaffirm the requirement for functionally assessing novel CASR variants prior to assigning causality to FHH.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
