{"title":"日本人群中排除 22q11.2 缺失综合征的动脉导管未闭的遗传学病因,以及 TMEM260 的流行变异 c.1617del 的鉴定。","authors":"Hisao Yaoita, Eiichiro Kawai, Jun Takayama, Shinya Iwasawa, Naoya Saijo, Masayuki Abiko, Kouta Suzuki, Masato Kimura, Akira Ozawa, Gen Tamiya, Shigeo Kure, Atsuo Kikuchi","doi":"10.1038/s10038-024-01223-y","DOIUrl":null,"url":null,"abstract":"Truncus Arteriosus (TA) is a congenital heart disease characterized by a single common blood vessel emerging from the right and left ventricles instead of the main pulmonary artery and aorta. TA accounts for 4% of all critical congenital heart diseases. The most common cause of TA is 22q11.2 deletion syndrome, accounting for 12–35% of all TA cases. However, no major causes of TA other than 22q11.2 deletion have been reported. We performed whole-genome sequencing of 11 Japanese patients having TA without 22q11.2 deletion. Among five patients, we identified pathogenic variants in TMEM260; the biallelic loss-of-function variants of which have recently been associated with structural heart defects and renal anomalies syndrome (SHDRA). In one patient, we identified a de novo pathogenic variant in GATA6, and in another patient, we identified a de novo probably pathogenic variant in NOTCH1. Notably, we identified a prevalent variant in TMEM260 (ENST00000261556.6), c.1617del (p.Trp539Cysfs*9), in 8/22 alleles among the 11 patients. The c.1617del variant was estimated to occur approximately 23 kiloyears ago. Based on the allele frequency of the c.1617del variant in the Japanese population (0.36%), approximately 26% of Japanese patients afflicted with TA could harbor homozygous c.1617del variants. This study highlights TMEM260, especially c.1617del, as a major genetic cause of TA in the Japanese population.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 5","pages":"177-183"},"PeriodicalIF":2.5000,"publicationDate":"2024-02-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s10038-024-01223-y.pdf","citationCount":"0","resultStr":"{\"title\":\"Genetic etiology of truncus arteriosus excluding 22q11.2 deletion syndrome and identification of c.1617del, a prevalent variant in TMEM260, in the Japanese population\",\"authors\":\"Hisao Yaoita, Eiichiro Kawai, Jun Takayama, Shinya Iwasawa, Naoya Saijo, Masayuki Abiko, Kouta Suzuki, Masato Kimura, Akira Ozawa, Gen Tamiya, Shigeo Kure, Atsuo Kikuchi\",\"doi\":\"10.1038/s10038-024-01223-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Truncus Arteriosus (TA) is a congenital heart disease characterized by a single common blood vessel emerging from the right and left ventricles instead of the main pulmonary artery and aorta. TA accounts for 4% of all critical congenital heart diseases. The most common cause of TA is 22q11.2 deletion syndrome, accounting for 12–35% of all TA cases. However, no major causes of TA other than 22q11.2 deletion have been reported. We performed whole-genome sequencing of 11 Japanese patients having TA without 22q11.2 deletion. Among five patients, we identified pathogenic variants in TMEM260; the biallelic loss-of-function variants of which have recently been associated with structural heart defects and renal anomalies syndrome (SHDRA). In one patient, we identified a de novo pathogenic variant in GATA6, and in another patient, we identified a de novo probably pathogenic variant in NOTCH1. Notably, we identified a prevalent variant in TMEM260 (ENST00000261556.6), c.1617del (p.Trp539Cysfs*9), in 8/22 alleles among the 11 patients. The c.1617del variant was estimated to occur approximately 23 kiloyears ago. Based on the allele frequency of the c.1617del variant in the Japanese population (0.36%), approximately 26% of Japanese patients afflicted with TA could harbor homozygous c.1617del variants. This study highlights TMEM260, especially c.1617del, as a major genetic cause of TA in the Japanese population.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 5\",\"pages\":\"177-183\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-02-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s10038-024-01223-y.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-024-01223-y\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01223-y","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Genetic etiology of truncus arteriosus excluding 22q11.2 deletion syndrome and identification of c.1617del, a prevalent variant in TMEM260, in the Japanese population
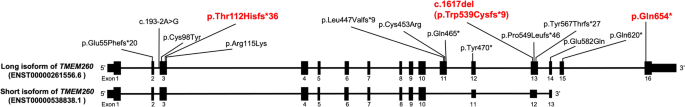
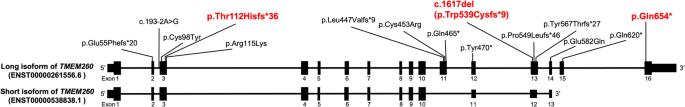
Truncus Arteriosus (TA) is a congenital heart disease characterized by a single common blood vessel emerging from the right and left ventricles instead of the main pulmonary artery and aorta. TA accounts for 4% of all critical congenital heart diseases. The most common cause of TA is 22q11.2 deletion syndrome, accounting for 12–35% of all TA cases. However, no major causes of TA other than 22q11.2 deletion have been reported. We performed whole-genome sequencing of 11 Japanese patients having TA without 22q11.2 deletion. Among five patients, we identified pathogenic variants in TMEM260; the biallelic loss-of-function variants of which have recently been associated with structural heart defects and renal anomalies syndrome (SHDRA). In one patient, we identified a de novo pathogenic variant in GATA6, and in another patient, we identified a de novo probably pathogenic variant in NOTCH1. Notably, we identified a prevalent variant in TMEM260 (ENST00000261556.6), c.1617del (p.Trp539Cysfs*9), in 8/22 alleles among the 11 patients. The c.1617del variant was estimated to occur approximately 23 kiloyears ago. Based on the allele frequency of the c.1617del variant in the Japanese population (0.36%), approximately 26% of Japanese patients afflicted with TA could harbor homozygous c.1617del variants. This study highlights TMEM260, especially c.1617del, as a major genetic cause of TA in the Japanese population.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
