{"title":"在一个患有短链烯酰-CoA 水合酶缺乏症和 Mt-DNA 缺失症的近亲家族中对新型 ECHS1 基因突变的分子和硅学研究:对三聚体组装和催化活性的影响。","authors":"Marwa Maalej, Lamia Sfaihi, Olfa-Alila Fersi, Boudour Khabou, Marwa Ammar, Rahma Felhi, Marwa Kharrat, Jihen Chouchen, Thouraya Kammoun, Abdelaziz Tlili, Faiza Fakhfakh","doi":"10.1007/s11011-024-01343-6","DOIUrl":null,"url":null,"abstract":"<p><p>Short-chain enoyl-CoA hydratase deficiency (ECHS1D) is a rare congenital metabolic disorder that follows an autosomal recessive inheritance pattern. It is caused by mutations in the ECHS1 gene, which encodes a mitochondrial enzyme involved in the second step of mitochondrial β-oxidation of fatty acids. The main characteristics of the disease are severe developmental delay, regression, seizures, neurodegeneration, high blood lactate, and a brain MRI pattern consistent with Leigh syndrome. Here, we report three patients belonging to a consanguineous family who presented with mitochondrial encephalomyopathy. Whole-exome sequencing revealed a new homozygous mutation c.619G > A (p.Gly207Ser) at the last nucleotide position in exon 5 of the ECHS1 gene. Experimental analysis showed that normal ECHS1 pre-mRNA splicing occurred in all patients compared to controls. Furthermore, three-dimensional models of wild-type and mutant echs1 proteins revealed changes in catalytic site interactions, conformational changes, and intramolecular interactions, potentially disrupting echs1 protein trimerization and affecting its function. Additionally, the quantification of mtDNA copy number variation in blood leukocytes showed severe mtDNA depletion in all probands.</p>","PeriodicalId":18685,"journal":{"name":"Metabolic brain disease","volume":" ","pages":"611-623"},"PeriodicalIF":3.5000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Molecular and in silico investigation of a novel ECHS1 gene mutation in a consanguine family with short-chain enoyl-CoA hydratase deficiency and Mt-DNA depletion: effect on trimer assembly and catalytic activity.\",\"authors\":\"Marwa Maalej, Lamia Sfaihi, Olfa-Alila Fersi, Boudour Khabou, Marwa Ammar, Rahma Felhi, Marwa Kharrat, Jihen Chouchen, Thouraya Kammoun, Abdelaziz Tlili, Faiza Fakhfakh\",\"doi\":\"10.1007/s11011-024-01343-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Short-chain enoyl-CoA hydratase deficiency (ECHS1D) is a rare congenital metabolic disorder that follows an autosomal recessive inheritance pattern. It is caused by mutations in the ECHS1 gene, which encodes a mitochondrial enzyme involved in the second step of mitochondrial β-oxidation of fatty acids. The main characteristics of the disease are severe developmental delay, regression, seizures, neurodegeneration, high blood lactate, and a brain MRI pattern consistent with Leigh syndrome. Here, we report three patients belonging to a consanguineous family who presented with mitochondrial encephalomyopathy. Whole-exome sequencing revealed a new homozygous mutation c.619G > A (p.Gly207Ser) at the last nucleotide position in exon 5 of the ECHS1 gene. Experimental analysis showed that normal ECHS1 pre-mRNA splicing occurred in all patients compared to controls. Furthermore, three-dimensional models of wild-type and mutant echs1 proteins revealed changes in catalytic site interactions, conformational changes, and intramolecular interactions, potentially disrupting echs1 protein trimerization and affecting its function. Additionally, the quantification of mtDNA copy number variation in blood leukocytes showed severe mtDNA depletion in all probands.</p>\",\"PeriodicalId\":18685,\"journal\":{\"name\":\"Metabolic brain disease\",\"volume\":\" \",\"pages\":\"611-623\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Metabolic brain disease\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s11011-024-01343-6\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/2/16 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Metabolic brain disease","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11011-024-01343-6","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/2/16 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Molecular and in silico investigation of a novel ECHS1 gene mutation in a consanguine family with short-chain enoyl-CoA hydratase deficiency and Mt-DNA depletion: effect on trimer assembly and catalytic activity.
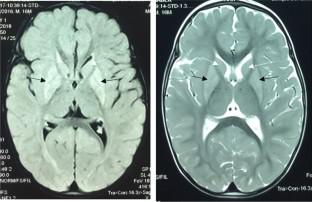
Short-chain enoyl-CoA hydratase deficiency (ECHS1D) is a rare congenital metabolic disorder that follows an autosomal recessive inheritance pattern. It is caused by mutations in the ECHS1 gene, which encodes a mitochondrial enzyme involved in the second step of mitochondrial β-oxidation of fatty acids. The main characteristics of the disease are severe developmental delay, regression, seizures, neurodegeneration, high blood lactate, and a brain MRI pattern consistent with Leigh syndrome. Here, we report three patients belonging to a consanguineous family who presented with mitochondrial encephalomyopathy. Whole-exome sequencing revealed a new homozygous mutation c.619G > A (p.Gly207Ser) at the last nucleotide position in exon 5 of the ECHS1 gene. Experimental analysis showed that normal ECHS1 pre-mRNA splicing occurred in all patients compared to controls. Furthermore, three-dimensional models of wild-type and mutant echs1 proteins revealed changes in catalytic site interactions, conformational changes, and intramolecular interactions, potentially disrupting echs1 protein trimerization and affecting its function. Additionally, the quantification of mtDNA copy number variation in blood leukocytes showed severe mtDNA depletion in all probands.
期刊介绍:
Metabolic Brain Disease serves as a forum for the publication of outstanding basic and clinical papers on all metabolic brain disease, including both human and animal studies. The journal publishes papers on the fundamental pathogenesis of these disorders and on related experimental and clinical techniques and methodologies. Metabolic Brain Disease is directed to physicians, neuroscientists, internists, psychiatrists, neurologists, pathologists, and others involved in the research and treatment of a broad range of metabolic brain disorders.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
