{"title":"E-pRSA:嵌入改进了蛋白质序列中残基相对溶剂可及性的预测","authors":"","doi":"10.1016/j.jmb.2024.168494","DOIUrl":null,"url":null,"abstract":"<div><p>Knowledge of the solvent accessibility of residues in a protein is essential for different applications, including the identification of interacting surfaces in protein–protein interactions and the characterization of variations. We describe E-pRSA, a novel web server to estimate Relative Solvent Accessibility values (RSAs) of residues directly from a protein sequence. The method exploits two complementary Protein Language Models to provide fast and accurate predictions. When benchmarked on different blind test sets, E-pRSA scores at the state-of-the-art, and outperforms a previous method we developed, DeepREx, which was based on sequence profiles after Multiple Sequence Alignments. The E-pRSA web server is freely available at <span><span>https://e-prsa.biocomp.unibo.it/main/</span><svg><path></path></svg></span> where users can submit single-sequence and batch jobs.</p></div>","PeriodicalId":369,"journal":{"name":"Journal of Molecular Biology","volume":"436 17","pages":"Article 168494"},"PeriodicalIF":4.7000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0022283624000664/pdfft?md5=5479e98c4394e85085ec9ab992a70ec7&pid=1-s2.0-S0022283624000664-main.pdf","citationCount":"0","resultStr":"{\"title\":\"E-pRSA: Embeddings Improve the Prediction of Residue Relative Solvent Accessibility in Protein Sequence\",\"authors\":\"\",\"doi\":\"10.1016/j.jmb.2024.168494\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Knowledge of the solvent accessibility of residues in a protein is essential for different applications, including the identification of interacting surfaces in protein–protein interactions and the characterization of variations. We describe E-pRSA, a novel web server to estimate Relative Solvent Accessibility values (RSAs) of residues directly from a protein sequence. The method exploits two complementary Protein Language Models to provide fast and accurate predictions. When benchmarked on different blind test sets, E-pRSA scores at the state-of-the-art, and outperforms a previous method we developed, DeepREx, which was based on sequence profiles after Multiple Sequence Alignments. The E-pRSA web server is freely available at <span><span>https://e-prsa.biocomp.unibo.it/main/</span><svg><path></path></svg></span> where users can submit single-sequence and batch jobs.</p></div>\",\"PeriodicalId\":369,\"journal\":{\"name\":\"Journal of Molecular Biology\",\"volume\":\"436 17\",\"pages\":\"Article 168494\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S0022283624000664/pdfft?md5=5479e98c4394e85085ec9ab992a70ec7&pid=1-s2.0-S0022283624000664-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0022283624000664\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022283624000664","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
E-pRSA: Embeddings Improve the Prediction of Residue Relative Solvent Accessibility in Protein Sequence
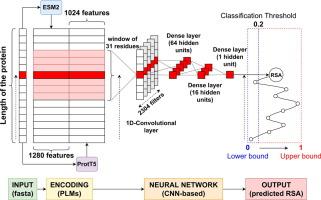
Knowledge of the solvent accessibility of residues in a protein is essential for different applications, including the identification of interacting surfaces in protein–protein interactions and the characterization of variations. We describe E-pRSA, a novel web server to estimate Relative Solvent Accessibility values (RSAs) of residues directly from a protein sequence. The method exploits two complementary Protein Language Models to provide fast and accurate predictions. When benchmarked on different blind test sets, E-pRSA scores at the state-of-the-art, and outperforms a previous method we developed, DeepREx, which was based on sequence profiles after Multiple Sequence Alignments. The E-pRSA web server is freely available at https://e-prsa.biocomp.unibo.it/main/ where users can submit single-sequence and batch jobs.
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
