{"title":"杂合子 CAPZA2 突变导致全面发育迟缓、肌张力低下和癫痫:病例报告和文献综述。","authors":"Xiao-Man Zhang, Kai-Li Xu, Jing-Hui Kong, Geng Dong, Shi-Jie Dong, Zhi-Xiao Yang, Shu-Jing Xu, Li Wang, Shu-Ying Luo, Yao-Dong Zhang, Chong-Chen Zhou, Wei-Yue Gu, Shi-Yue Mei","doi":"10.1038/s10038-024-01230-z","DOIUrl":null,"url":null,"abstract":"CAPZA2 encodes the α2 subunit of CAPZA, which is vital for actin polymerization and depolymerization in humans. However, understanding of diseases associated with CAPZA2 remains limited. To date, only three cases have been documented with neurodevelopmental abnormalities such as delayed motor development, speech delay, intellectual disability, hypotonia, and a history of seizures. In this study, we document a patient who exhibited seizures, mild intellectual disability, and impaired motor development yet did not demonstrate speech delay or hypotonia. The patient also suffered from recurrent instances of respiratory infections, gastrointestinal and allergic diseases. A novel de novo splicing variant c.219+1 G > A was detected in the CAPZA2 gene through whole-exome sequencing. This variant led to exon 4 skipping in mRNA splicing, confirmed by RT-PCR and Sanger sequencing. To our knowledge, this is the third study on human CAPZA2 defects, documenting the fourth unambiguously diagnosed case. Furthermore, this splicing mutation type is reported here for the first time. Our research offers additional support for the existence of a CAPZA2-related non-syndromic neurodevelopmental disorder. Our findings augment our understanding of the phenotypic range associated with CAPZA2 deficiency and enrich the knowledge of the mutational spectrum of the CAPZA2 gene.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 5","pages":"197-203"},"PeriodicalIF":2.5000,"publicationDate":"2024-02-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Heterozygous CAPZA2 mutations cause global developmental delay, hypotonia with epilepsy: a case report and the literature review\",\"authors\":\"Xiao-Man Zhang, Kai-Li Xu, Jing-Hui Kong, Geng Dong, Shi-Jie Dong, Zhi-Xiao Yang, Shu-Jing Xu, Li Wang, Shu-Ying Luo, Yao-Dong Zhang, Chong-Chen Zhou, Wei-Yue Gu, Shi-Yue Mei\",\"doi\":\"10.1038/s10038-024-01230-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"CAPZA2 encodes the α2 subunit of CAPZA, which is vital for actin polymerization and depolymerization in humans. However, understanding of diseases associated with CAPZA2 remains limited. To date, only three cases have been documented with neurodevelopmental abnormalities such as delayed motor development, speech delay, intellectual disability, hypotonia, and a history of seizures. In this study, we document a patient who exhibited seizures, mild intellectual disability, and impaired motor development yet did not demonstrate speech delay or hypotonia. The patient also suffered from recurrent instances of respiratory infections, gastrointestinal and allergic diseases. A novel de novo splicing variant c.219+1 G > A was detected in the CAPZA2 gene through whole-exome sequencing. This variant led to exon 4 skipping in mRNA splicing, confirmed by RT-PCR and Sanger sequencing. To our knowledge, this is the third study on human CAPZA2 defects, documenting the fourth unambiguously diagnosed case. Furthermore, this splicing mutation type is reported here for the first time. Our research offers additional support for the existence of a CAPZA2-related non-syndromic neurodevelopmental disorder. Our findings augment our understanding of the phenotypic range associated with CAPZA2 deficiency and enrich the knowledge of the mutational spectrum of the CAPZA2 gene.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 5\",\"pages\":\"197-203\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-02-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-024-01230-z\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01230-z","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Heterozygous CAPZA2 mutations cause global developmental delay, hypotonia with epilepsy: a case report and the literature review
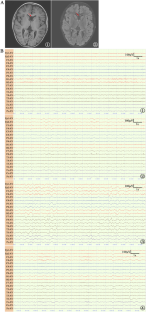
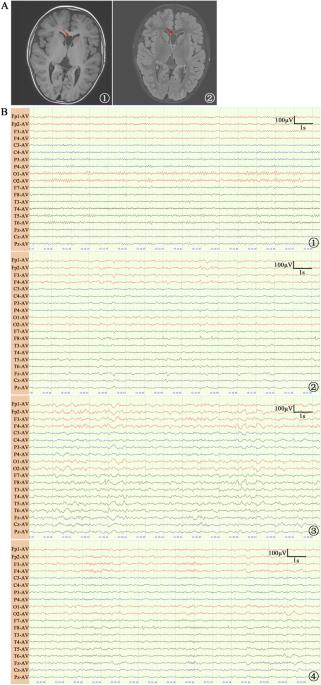
CAPZA2 encodes the α2 subunit of CAPZA, which is vital for actin polymerization and depolymerization in humans. However, understanding of diseases associated with CAPZA2 remains limited. To date, only three cases have been documented with neurodevelopmental abnormalities such as delayed motor development, speech delay, intellectual disability, hypotonia, and a history of seizures. In this study, we document a patient who exhibited seizures, mild intellectual disability, and impaired motor development yet did not demonstrate speech delay or hypotonia. The patient also suffered from recurrent instances of respiratory infections, gastrointestinal and allergic diseases. A novel de novo splicing variant c.219+1 G > A was detected in the CAPZA2 gene through whole-exome sequencing. This variant led to exon 4 skipping in mRNA splicing, confirmed by RT-PCR and Sanger sequencing. To our knowledge, this is the third study on human CAPZA2 defects, documenting the fourth unambiguously diagnosed case. Furthermore, this splicing mutation type is reported here for the first time. Our research offers additional support for the existence of a CAPZA2-related non-syndromic neurodevelopmental disorder. Our findings augment our understanding of the phenotypic range associated with CAPZA2 deficiency and enrich the knowledge of the mutational spectrum of the CAPZA2 gene.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
