Paniz Farshadyeganeh, Takahiro Yamada, Hirofumi Ohashi, Gen Nishimura, Hiroki Fujita, Yuriko Oishi, Misa Nunode, Shuku Ishikawa, Jun Murotsuki, Yuri Yamashita, Shiro Ikegawa, Tomoo Ogi, Eri Arikawa-Hirasawa, Kinji Ohno
{"title":"Rolland-Desbuquois型发育不良是由HSPG2的致病变体引起的,这是五名患者共有的一个创始单倍型。","authors":"Paniz Farshadyeganeh, Takahiro Yamada, Hirofumi Ohashi, Gen Nishimura, Hiroki Fujita, Yuriko Oishi, Misa Nunode, Shuku Ishikawa, Jun Murotsuki, Yuri Yamashita, Shiro Ikegawa, Tomoo Ogi, Eri Arikawa-Hirasawa, Kinji Ohno","doi":"10.1038/s10038-024-01229-6","DOIUrl":null,"url":null,"abstract":"Dyssegmental dysplasia (DD) is a severe skeletal dysplasia comprised of two subtypes: lethal Silverman–Handmaker type (DDSH) and nonlethal Rolland–Desbuquois type (DDRD). DDSH is caused by biallelic pathogenic variants in HSPG2 encoding perlecan, whereas the genetic cause of DDRD remains undetermined. Schwartz–Jampel syndrome (SJS) is also caused by biallelic pathogenic variants in HSPG2 and is an allelic disorder of DDSH. In SJS and DDSH, 44 and 8 pathogenic variants have been reported in HSPG2, respectively. Here, we report that five patients with DDRD carried four pathogenic variants in HSPG2: c.9970 G > A (p.G3324R), c.559 C > T (p.R187X), c7006 + 1 G > A, and c.11562 + 2 T > G. Two patients were homozygous for p.G3324R, and three patients were heterozygous for p.G3324R. Haplotype analysis revealed a founder haplotype spanning 85,973 bp shared in the five patients. SJS, DDRD, and DDSH are allelic disorders with pathogenic variants in HSPG2.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 6","pages":"235-244"},"PeriodicalIF":2.3000,"publicationDate":"2024-02-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s10038-024-01229-6.pdf","citationCount":"0","resultStr":"{\"title\":\"Dyssegmental dysplasia Rolland–Desbuquois type is caused by pathogenic variants in HSPG2 - a founder haplotype shared in five patients\",\"authors\":\"Paniz Farshadyeganeh, Takahiro Yamada, Hirofumi Ohashi, Gen Nishimura, Hiroki Fujita, Yuriko Oishi, Misa Nunode, Shuku Ishikawa, Jun Murotsuki, Yuri Yamashita, Shiro Ikegawa, Tomoo Ogi, Eri Arikawa-Hirasawa, Kinji Ohno\",\"doi\":\"10.1038/s10038-024-01229-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Dyssegmental dysplasia (DD) is a severe skeletal dysplasia comprised of two subtypes: lethal Silverman–Handmaker type (DDSH) and nonlethal Rolland–Desbuquois type (DDRD). DDSH is caused by biallelic pathogenic variants in HSPG2 encoding perlecan, whereas the genetic cause of DDRD remains undetermined. Schwartz–Jampel syndrome (SJS) is also caused by biallelic pathogenic variants in HSPG2 and is an allelic disorder of DDSH. In SJS and DDSH, 44 and 8 pathogenic variants have been reported in HSPG2, respectively. Here, we report that five patients with DDRD carried four pathogenic variants in HSPG2: c.9970 G > A (p.G3324R), c.559 C > T (p.R187X), c7006 + 1 G > A, and c.11562 + 2 T > G. Two patients were homozygous for p.G3324R, and three patients were heterozygous for p.G3324R. Haplotype analysis revealed a founder haplotype spanning 85,973 bp shared in the five patients. SJS, DDRD, and DDSH are allelic disorders with pathogenic variants in HSPG2.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 6\",\"pages\":\"235-244\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2024-02-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s10038-024-01229-6.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-024-01229-6\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01229-6","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
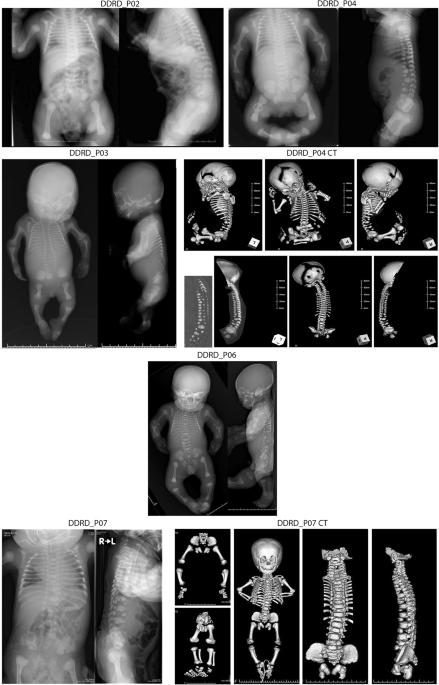
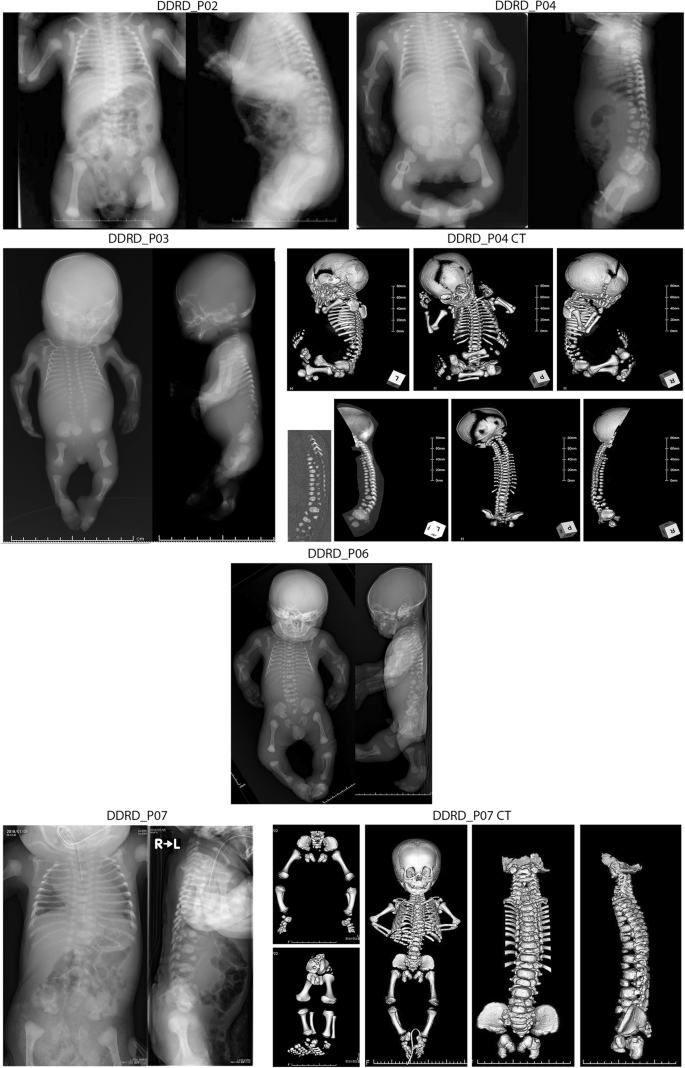
骨骼发育不良(DD)是一种严重的骨骼发育不良,包括两个亚型:致死型 Silverman-Handmaker 型(DDSH)和非致死型 Rolland-Desbuquois 型(DDRD)。DDSH是由编码perlecan的HSPG2的双偶性致病变体引起的,而DDRD的遗传原因仍未确定。Schwartz-Jampel 综合征(SJS)也是由 HSPG2 的双等位致病变体引起的,是 DDSH 的等位基因紊乱。在 SJS 和 DDSH 中,HSPG2 中分别有 44 个和 8 个致病变体。在此,我们报告了五名 DDRD 患者携带 HSPG2 中的四个致病变体:c.9970 G > A (p.G3324R)、c.559 C > T (p.R187X)、c7006 + 1 G > A 和 c.11562 + 2 T > G。单倍型分析显示,五名患者共有一个跨度为 85,973 bp 的创始单倍型。SJS、DDRD 和 DDSH 是具有 HSPG2 致病变体的等位基因疾病。
Dyssegmental dysplasia Rolland–Desbuquois type is caused by pathogenic variants in HSPG2 - a founder haplotype shared in five patients
Dyssegmental dysplasia (DD) is a severe skeletal dysplasia comprised of two subtypes: lethal Silverman–Handmaker type (DDSH) and nonlethal Rolland–Desbuquois type (DDRD). DDSH is caused by biallelic pathogenic variants in HSPG2 encoding perlecan, whereas the genetic cause of DDRD remains undetermined. Schwartz–Jampel syndrome (SJS) is also caused by biallelic pathogenic variants in HSPG2 and is an allelic disorder of DDSH. In SJS and DDSH, 44 and 8 pathogenic variants have been reported in HSPG2, respectively. Here, we report that five patients with DDRD carried four pathogenic variants in HSPG2: c.9970 G > A (p.G3324R), c.559 C > T (p.R187X), c7006 + 1 G > A, and c.11562 + 2 T > G. Two patients were homozygous for p.G3324R, and three patients were heterozygous for p.G3324R. Haplotype analysis revealed a founder haplotype spanning 85,973 bp shared in the five patients. SJS, DDRD, and DDSH are allelic disorders with pathogenic variants in HSPG2.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
