{"title":"金属-金属五键的理论光电子能谱学:相对论驱动的前沿轨道重排","authors":"Abhik Ghosh*, and , Jeanet Conradie*, ","doi":"10.1021/acsorginorgau.4c00002","DOIUrl":null,"url":null,"abstract":"<p >A recent reinvestigation of the gas-phase photoelectron spectra of Group 6 metal–metal quadruple-bonded complexes with scalar-relativistic DFT calculations showed that common exchange-correlation functionals reproduce the lowest ionization potentials in a semiquantitative manner. The finding encouraged us to undertake a DFT study of metal–metal quintuple bonds in a set of bisamidinato complexes with the formula M<sup>I</sup><sub>2</sub>[HC(NR)<sub>2</sub>]<sub>2</sub> (M = Cr, Mo, W; R = H, Ph, 2,6-<i>i</i>Pr<sub>2</sub>C<sub>6</sub>H<sub>3</sub>) and idealized <i>D</i><sub>2<i>h</i></sub> symmetry. Scalar-relativistic OLYP/STO-TZ2P calculations indicated significant shifts in valence orbital energies among the three metals, which translate to lower first ionization potentials, higher electron affinities, and lower HOMO–LUMO gaps for the W complexes relative to their Cr and Mo counterparts. These differences are largely attributable to substantially larger relativistic effects in the case of tungsten relative to those of its lighter congeners.</p>","PeriodicalId":29797,"journal":{"name":"ACS Organic & Inorganic Au","volume":"4 3","pages":"301–305"},"PeriodicalIF":3.3000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acsorginorgau.4c00002","citationCount":"0","resultStr":"{\"title\":\"Theoretical Photoelectron Spectroscopy of Metal–Metal Quintuple Bonds: Relativity-Driven Reordering of Frontier Orbitals\",\"authors\":\"Abhik Ghosh*, and , Jeanet Conradie*, \",\"doi\":\"10.1021/acsorginorgau.4c00002\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >A recent reinvestigation of the gas-phase photoelectron spectra of Group 6 metal–metal quadruple-bonded complexes with scalar-relativistic DFT calculations showed that common exchange-correlation functionals reproduce the lowest ionization potentials in a semiquantitative manner. The finding encouraged us to undertake a DFT study of metal–metal quintuple bonds in a set of bisamidinato complexes with the formula M<sup>I</sup><sub>2</sub>[HC(NR)<sub>2</sub>]<sub>2</sub> (M = Cr, Mo, W; R = H, Ph, 2,6-<i>i</i>Pr<sub>2</sub>C<sub>6</sub>H<sub>3</sub>) and idealized <i>D</i><sub>2<i>h</i></sub> symmetry. Scalar-relativistic OLYP/STO-TZ2P calculations indicated significant shifts in valence orbital energies among the three metals, which translate to lower first ionization potentials, higher electron affinities, and lower HOMO–LUMO gaps for the W complexes relative to their Cr and Mo counterparts. These differences are largely attributable to substantially larger relativistic effects in the case of tungsten relative to those of its lighter congeners.</p>\",\"PeriodicalId\":29797,\"journal\":{\"name\":\"ACS Organic & Inorganic Au\",\"volume\":\"4 3\",\"pages\":\"301–305\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acsorginorgau.4c00002\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Organic & Inorganic Au\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acsorginorgau.4c00002\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Organic & Inorganic Au","FirstCategoryId":"1085","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsorginorgau.4c00002","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Theoretical Photoelectron Spectroscopy of Metal–Metal Quintuple Bonds: Relativity-Driven Reordering of Frontier Orbitals
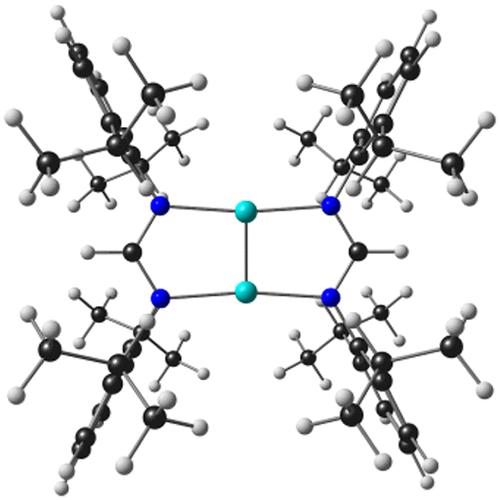
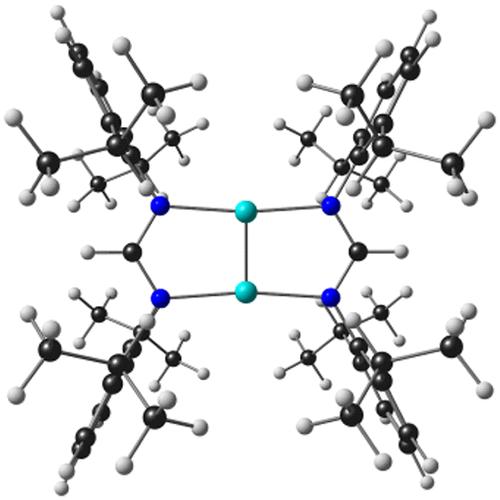
A recent reinvestigation of the gas-phase photoelectron spectra of Group 6 metal–metal quadruple-bonded complexes with scalar-relativistic DFT calculations showed that common exchange-correlation functionals reproduce the lowest ionization potentials in a semiquantitative manner. The finding encouraged us to undertake a DFT study of metal–metal quintuple bonds in a set of bisamidinato complexes with the formula MI2[HC(NR)2]2 (M = Cr, Mo, W; R = H, Ph, 2,6-iPr2C6H3) and idealized D2h symmetry. Scalar-relativistic OLYP/STO-TZ2P calculations indicated significant shifts in valence orbital energies among the three metals, which translate to lower first ionization potentials, higher electron affinities, and lower HOMO–LUMO gaps for the W complexes relative to their Cr and Mo counterparts. These differences are largely attributable to substantially larger relativistic effects in the case of tungsten relative to those of its lighter congeners.
期刊介绍:
ACS Organic & Inorganic Au is an open access journal that publishes original experimental and theoretical/computational studies on organic organometallic inorganic crystal growth and engineering and organic process chemistry. Short letters comprehensive articles reviews and perspectives are welcome on topics that include:Organic chemistry Organometallic chemistry Inorganic Chemistry and Organic Process Chemistry.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
