{"title":"对一名因 FRMPD4 蛋白的 FERM 结构域中的新型变异而导致严重智力障碍的儿童的遗传分析","authors":"Hua Pan, Feng Zhu, Kun Chen, Yin Zhang","doi":"10.1007/s12041-024-01465-x","DOIUrl":null,"url":null,"abstract":"<p>Intellectual developmental disorder, X-linked 104 (XLID104), caused by the <i>FRMPD4</i> gene variant, is a rare X-linked genetic disease that primarily manifests as intellectual disability (ID) and language delay, and may be accompanied by behavioural abnormalities. Currently, only 11 patients from four families have been reported to carry <i>FRMPD4</i> gene variants. Here, we report a rare case of a Chinese patient with XLID104 who was presented with severe ID and language impairment. Genetic testing results showed that the patient had a novel hemizygous variant on <i>FRMPD4</i> inherited from the heterozygous variant NM_001368397: c.1772A>C (p.Glu591Ala) carried by his mother. To our knowledge, this variant has not been reported previously. Western blot results for the recombinant plasmid constructed <i>in vitro</i> indicated that the expression of the mutant protein may be reduced. Using molecular dynamics simulations, we predicted that the mutant protein may affect the interaction of the FRMPD4 protein with DLG4. In this study, we expand the spectrum of <i>FRMPD4</i> variants and suggest that the clinical awareness of the genetic diagnosis of nonsyndromic ID should be strengthened.</p>","PeriodicalId":15907,"journal":{"name":"Journal of Genetics","volume":"21 1","pages":""},"PeriodicalIF":1.2000,"publicationDate":"2024-03-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Genetic analysis of a child with severe intellectual disability caused by a novel variant in the FERM domain of the FRMPD4 protein\",\"authors\":\"Hua Pan, Feng Zhu, Kun Chen, Yin Zhang\",\"doi\":\"10.1007/s12041-024-01465-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Intellectual developmental disorder, X-linked 104 (XLID104), caused by the <i>FRMPD4</i> gene variant, is a rare X-linked genetic disease that primarily manifests as intellectual disability (ID) and language delay, and may be accompanied by behavioural abnormalities. Currently, only 11 patients from four families have been reported to carry <i>FRMPD4</i> gene variants. Here, we report a rare case of a Chinese patient with XLID104 who was presented with severe ID and language impairment. Genetic testing results showed that the patient had a novel hemizygous variant on <i>FRMPD4</i> inherited from the heterozygous variant NM_001368397: c.1772A>C (p.Glu591Ala) carried by his mother. To our knowledge, this variant has not been reported previously. Western blot results for the recombinant plasmid constructed <i>in vitro</i> indicated that the expression of the mutant protein may be reduced. Using molecular dynamics simulations, we predicted that the mutant protein may affect the interaction of the FRMPD4 protein with DLG4. In this study, we expand the spectrum of <i>FRMPD4</i> variants and suggest that the clinical awareness of the genetic diagnosis of nonsyndromic ID should be strengthened.</p>\",\"PeriodicalId\":15907,\"journal\":{\"name\":\"Journal of Genetics\",\"volume\":\"21 1\",\"pages\":\"\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2024-03-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s12041-024-01465-x\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"EDUCATION & EDUCATIONAL RESEARCH\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s12041-024-01465-x","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EDUCATION & EDUCATIONAL RESEARCH","Score":null,"Total":0}
引用次数: 0
摘要
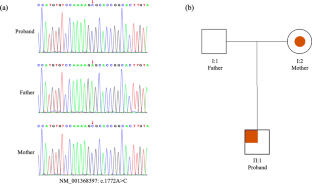
由 FRMPD4 基因变异引起的 X 连锁智力发育障碍 104(XLID104)是一种罕见的 X 连锁遗传病,主要表现为智力障碍(ID)和语言发育迟缓,并可能伴有行为异常。目前,仅有四个家族的11名患者被报道携带FRMPD4基因变异。在此,我们报告了一例罕见的中国 XLID104 患者,该患者表现为严重的智障和语言障碍。基因检测结果显示,该患者的 FRMPD4 基因存在一个新的半杂合子变异,该变异遗传自其母亲携带的杂合子变异 NM_001368397:c.1772A>C (p.Glu591Ala)。据我们所知,这种变异以前从未报道过。体外构建的重组质粒的 Western 印迹结果表明,突变体蛋白的表达可能会降低。通过分子动力学模拟,我们预测突变体蛋白可能会影响 FRMPD4 蛋白与 DLG4 的相互作用。在这项研究中,我们扩大了 FRMPD4 变异的范围,并建议临床上应加强对非综合征 ID 基因诊断的认识。
Genetic analysis of a child with severe intellectual disability caused by a novel variant in the FERM domain of the FRMPD4 protein
Intellectual developmental disorder, X-linked 104 (XLID104), caused by the FRMPD4 gene variant, is a rare X-linked genetic disease that primarily manifests as intellectual disability (ID) and language delay, and may be accompanied by behavioural abnormalities. Currently, only 11 patients from four families have been reported to carry FRMPD4 gene variants. Here, we report a rare case of a Chinese patient with XLID104 who was presented with severe ID and language impairment. Genetic testing results showed that the patient had a novel hemizygous variant on FRMPD4 inherited from the heterozygous variant NM_001368397: c.1772A>C (p.Glu591Ala) carried by his mother. To our knowledge, this variant has not been reported previously. Western blot results for the recombinant plasmid constructed in vitro indicated that the expression of the mutant protein may be reduced. Using molecular dynamics simulations, we predicted that the mutant protein may affect the interaction of the FRMPD4 protein with DLG4. In this study, we expand the spectrum of FRMPD4 variants and suggest that the clinical awareness of the genetic diagnosis of nonsyndromic ID should be strengthened.
期刊介绍:
The journal retains its traditional interest in evolutionary research that is of relevance to geneticists, even if this is not explicitly genetical in nature. The journal covers all areas of genetics and evolution,including molecular genetics and molecular evolution.It publishes papers and review articles on current topics, commentaries and essayson ideas and trends in genetics and evolutionary biology, historical developments, debates and book reviews. From 2010 onwards, the journal has published a special category of papers termed ‘Online Resources’. These are brief reports on the development and the routine use of molecular markers for assessing genetic variability within and among species. Also published are reports outlining pedagogical approaches in genetics teaching.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
