Shelby E Redfield, Pedro De-la-Torre, Mina Zamani, Hanjun Wang, Hina Khan, Tyler Morris, Gholamreza Shariati, Majid Karimi, Margaret A Kenna, Go Hun Seo, Hongen Xu, Wei Lu, Sadaf Naz, Hamid Galehdari, Artur A Indzhykulian, A Eliot Shearer, Barbara Vona
{"title":"PKHD1L1是一种参与立体纤毛外衣的基因,可导致常染色体隐性非综合征性听力损失。","authors":"Shelby E Redfield, Pedro De-la-Torre, Mina Zamani, Hanjun Wang, Hina Khan, Tyler Morris, Gholamreza Shariati, Majid Karimi, Margaret A Kenna, Go Hun Seo, Hongen Xu, Wei Lu, Sadaf Naz, Hamid Galehdari, Artur A Indzhykulian, A Eliot Shearer, Barbara Vona","doi":"10.1007/s00439-024-02649-2","DOIUrl":null,"url":null,"abstract":"<p><p>Identification of genes associated with nonsyndromic hearing loss is a crucial endeavor given the substantial number of individuals who remain without a diagnosis after even the most advanced genetic testing. PKHD1L1 was established as necessary for the formation of the cochlear hair-cell stereociliary coat and causes hearing loss in mice and zebrafish when mutated. We sought to determine if biallelic variants in PKHD1L1 also cause hearing loss in humans. Exome sequencing was performed on DNA of four families segregating autosomal recessive nonsyndromic sensorineural hearing loss. Compound heterozygous p.[(Gly129Ser)];p.[(Gly1314Val)] and p.[(Gly605Arg)];p[(Leu2818TyrfsTer5)], homozygous missense p.(His2479Gln) and nonsense p.(Arg3381Ter) variants were identified in PKHD1L1 that were predicted to be damaging using in silico pathogenicity prediction methods. In vitro functional analysis of two missense variants was performed using purified recombinant PKHD1L1 protein fragments. We then evaluated protein thermodynamic stability with and without the missense variants found in one of the families and performed a minigene splicing assay for another variant. In silico molecular modeling using AlphaFold2 and protein sequence alignment analysis were carried out to further explore potential variant effects on structure. In vitro functional assessment indicated that both engineered PKHD1L1 p.(Gly129Ser) and p.(Gly1314Val) mutant constructs significantly reduced the folding and structural stabilities of the expressed protein fragments, providing further evidence to support pathogenicity of these variants. Minigene assay of the c.1813G>A p.(Gly605Arg) variant, located at the boundary of exon 17, revealed exon skipping leading to an in-frame deletion of 48 amino acids. In silico molecular modeling exposed key structural features that might suggest PKHD1L1 protein destabilization. Multiple lines of evidence collectively associate PKHD1L1 with nonsyndromic mild-moderate to severe sensorineural hearing loss. PKHD1L1 testing in individuals with mild-moderate hearing loss may identify further affected families.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"311-329"},"PeriodicalIF":3.8000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11043200/pdf/","citationCount":"0","resultStr":"{\"title\":\"PKHD1L1, a gene involved in the stereocilia coat, causes autosomal recessive nonsyndromic hearing loss.\",\"authors\":\"Shelby E Redfield, Pedro De-la-Torre, Mina Zamani, Hanjun Wang, Hina Khan, Tyler Morris, Gholamreza Shariati, Majid Karimi, Margaret A Kenna, Go Hun Seo, Hongen Xu, Wei Lu, Sadaf Naz, Hamid Galehdari, Artur A Indzhykulian, A Eliot Shearer, Barbara Vona\",\"doi\":\"10.1007/s00439-024-02649-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Identification of genes associated with nonsyndromic hearing loss is a crucial endeavor given the substantial number of individuals who remain without a diagnosis after even the most advanced genetic testing. PKHD1L1 was established as necessary for the formation of the cochlear hair-cell stereociliary coat and causes hearing loss in mice and zebrafish when mutated. We sought to determine if biallelic variants in PKHD1L1 also cause hearing loss in humans. Exome sequencing was performed on DNA of four families segregating autosomal recessive nonsyndromic sensorineural hearing loss. Compound heterozygous p.[(Gly129Ser)];p.[(Gly1314Val)] and p.[(Gly605Arg)];p[(Leu2818TyrfsTer5)], homozygous missense p.(His2479Gln) and nonsense p.(Arg3381Ter) variants were identified in PKHD1L1 that were predicted to be damaging using in silico pathogenicity prediction methods. In vitro functional analysis of two missense variants was performed using purified recombinant PKHD1L1 protein fragments. We then evaluated protein thermodynamic stability with and without the missense variants found in one of the families and performed a minigene splicing assay for another variant. In silico molecular modeling using AlphaFold2 and protein sequence alignment analysis were carried out to further explore potential variant effects on structure. In vitro functional assessment indicated that both engineered PKHD1L1 p.(Gly129Ser) and p.(Gly1314Val) mutant constructs significantly reduced the folding and structural stabilities of the expressed protein fragments, providing further evidence to support pathogenicity of these variants. Minigene assay of the c.1813G>A p.(Gly605Arg) variant, located at the boundary of exon 17, revealed exon skipping leading to an in-frame deletion of 48 amino acids. In silico molecular modeling exposed key structural features that might suggest PKHD1L1 protein destabilization. Multiple lines of evidence collectively associate PKHD1L1 with nonsyndromic mild-moderate to severe sensorineural hearing loss. PKHD1L1 testing in individuals with mild-moderate hearing loss may identify further affected families.</p>\",\"PeriodicalId\":13175,\"journal\":{\"name\":\"Human Genetics\",\"volume\":\" \",\"pages\":\"311-329\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2024-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11043200/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s00439-024-02649-2\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/3/9 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02649-2","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/9 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
PKHD1L1, a gene involved in the stereocilia coat, causes autosomal recessive nonsyndromic hearing loss.
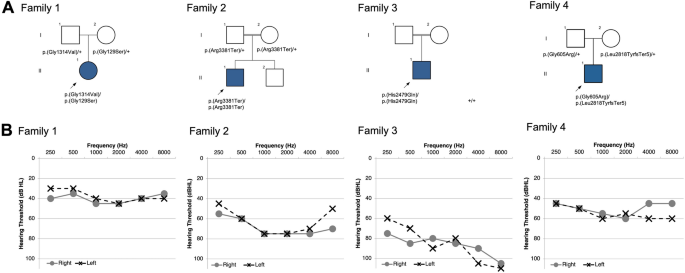
Identification of genes associated with nonsyndromic hearing loss is a crucial endeavor given the substantial number of individuals who remain without a diagnosis after even the most advanced genetic testing. PKHD1L1 was established as necessary for the formation of the cochlear hair-cell stereociliary coat and causes hearing loss in mice and zebrafish when mutated. We sought to determine if biallelic variants in PKHD1L1 also cause hearing loss in humans. Exome sequencing was performed on DNA of four families segregating autosomal recessive nonsyndromic sensorineural hearing loss. Compound heterozygous p.[(Gly129Ser)];p.[(Gly1314Val)] and p.[(Gly605Arg)];p[(Leu2818TyrfsTer5)], homozygous missense p.(His2479Gln) and nonsense p.(Arg3381Ter) variants were identified in PKHD1L1 that were predicted to be damaging using in silico pathogenicity prediction methods. In vitro functional analysis of two missense variants was performed using purified recombinant PKHD1L1 protein fragments. We then evaluated protein thermodynamic stability with and without the missense variants found in one of the families and performed a minigene splicing assay for another variant. In silico molecular modeling using AlphaFold2 and protein sequence alignment analysis were carried out to further explore potential variant effects on structure. In vitro functional assessment indicated that both engineered PKHD1L1 p.(Gly129Ser) and p.(Gly1314Val) mutant constructs significantly reduced the folding and structural stabilities of the expressed protein fragments, providing further evidence to support pathogenicity of these variants. Minigene assay of the c.1813G>A p.(Gly605Arg) variant, located at the boundary of exon 17, revealed exon skipping leading to an in-frame deletion of 48 amino acids. In silico molecular modeling exposed key structural features that might suggest PKHD1L1 protein destabilization. Multiple lines of evidence collectively associate PKHD1L1 with nonsyndromic mild-moderate to severe sensorineural hearing loss. PKHD1L1 testing in individuals with mild-moderate hearing loss may identify further affected families.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
