{"title":"对与 MADD 相关的神经发育障碍的临床和分子谱的新认识。","authors":"Ghada M. H. Abdel-Salam, Mohamed S. Abdel-Hamid","doi":"10.1038/s10038-024-01236-7","DOIUrl":null,"url":null,"abstract":"Biallelic pathogenic variants in MADD lead to a very rare neurodevelopmental disorder which is phenotypically pleiotropic grossly ranging from severe neonatal hypotonia, failure to thrive, multiple organ dysfunction, and early lethality to a similar but milder phenotype with better survival. Here, we report 5 patients from 3 unrelated Egyptian families in whom 4 patients showed the severe end of the spectrum displaying neonatal respiratory distress, hypotonia and chronic diarrhea while one patient presented with the mild form displaying moderate intellectual disability and myopathy. In addition, we observed distal arthrogryposis and nonspecific structural brain anomalies in all our patients. Interestingly, cerebellar and brainstem hypoplasia were noted in one patient. Whole exome sequencing identified three novel homozygous variants in the MADD gene: two likely pathogenic [c.4321delC p.(Gln1441ArgfsTer46) and c.2620 C > T p.(Arg874Ter)] and one variant of uncertain significance (c.4307 G > A, p.Arg1436Gln). The variants segregated with the disease in all available family members. Our findings confirm that arthrogryposis, genital, cardiac and structural brain anomalies are manifestations of MADD which expand the spectrum of MADD-related neurodevelopmental disorder. Moreover, they further highlight the convergence of MADD variants on different organ systems leading to complex phenotypes.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 6","pages":"263-270"},"PeriodicalIF":2.5000,"publicationDate":"2024-03-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s10038-024-01236-7.pdf","citationCount":"0","resultStr":"{\"title\":\"New insights into the clinical and molecular spectrum of the MADD-related neurodevelopmental disorder\",\"authors\":\"Ghada M. H. Abdel-Salam, Mohamed S. Abdel-Hamid\",\"doi\":\"10.1038/s10038-024-01236-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Biallelic pathogenic variants in MADD lead to a very rare neurodevelopmental disorder which is phenotypically pleiotropic grossly ranging from severe neonatal hypotonia, failure to thrive, multiple organ dysfunction, and early lethality to a similar but milder phenotype with better survival. Here, we report 5 patients from 3 unrelated Egyptian families in whom 4 patients showed the severe end of the spectrum displaying neonatal respiratory distress, hypotonia and chronic diarrhea while one patient presented with the mild form displaying moderate intellectual disability and myopathy. In addition, we observed distal arthrogryposis and nonspecific structural brain anomalies in all our patients. Interestingly, cerebellar and brainstem hypoplasia were noted in one patient. Whole exome sequencing identified three novel homozygous variants in the MADD gene: two likely pathogenic [c.4321delC p.(Gln1441ArgfsTer46) and c.2620 C > T p.(Arg874Ter)] and one variant of uncertain significance (c.4307 G > A, p.Arg1436Gln). The variants segregated with the disease in all available family members. Our findings confirm that arthrogryposis, genital, cardiac and structural brain anomalies are manifestations of MADD which expand the spectrum of MADD-related neurodevelopmental disorder. Moreover, they further highlight the convergence of MADD variants on different organ systems leading to complex phenotypes.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 6\",\"pages\":\"263-270\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-03-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s10038-024-01236-7.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-024-01236-7\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01236-7","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
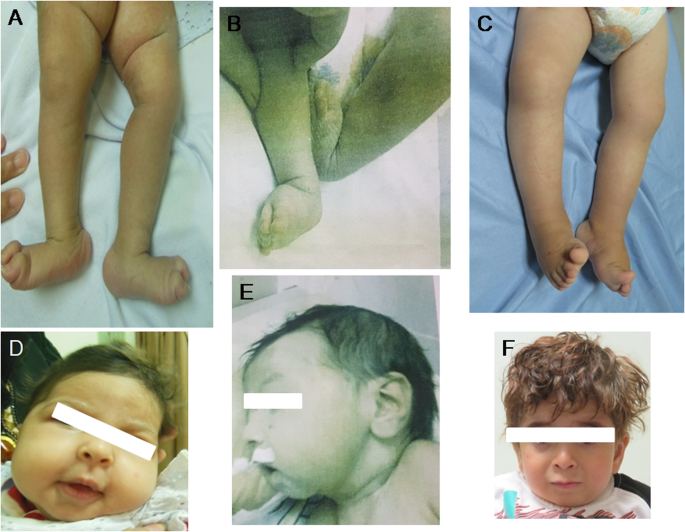
MADD的双倍拷贝致病变异会导致一种非常罕见的神经发育障碍,其表型具有多重性,从严重的新生儿肌张力低下、发育不良、多器官功能障碍和早期致死,到表型相似但病情较轻且存活率较高。在本文中,我们报告了来自3个无血缘关系的埃及家庭的5名患者,其中4名患者表现为重度,表现为新生儿呼吸窘迫、肌张力低下和慢性腹泻,1名患者表现为轻度,表现为中度智力障碍和肌病。此外,我们还在所有患者中观察到远端关节畸形和非特异性脑结构异常。有趣的是,其中一名患者出现了小脑和脑干发育不全。全外显子组测序在 MADD 基因中发现了三个新的同源变异:两个可能致病的变异 [c.4321delC p.(Gln1441ArgfsTer46) 和 c.2620 C > T p.(Arg874Ter)] 以及一个意义不明的变异(c.4307 G > A, p.Arg1436Gln)。这些变异与所有家族成员的疾病都有分离。我们的研究结果证实,关节畸形、生殖器、心脏和脑结构异常是 MADD 的表现形式,扩大了 MADD 相关神经发育障碍的范围。此外,这些研究还进一步突显了 MADD 变异在不同器官系统中的汇聚导致了复杂的表型。
New insights into the clinical and molecular spectrum of the MADD-related neurodevelopmental disorder
Biallelic pathogenic variants in MADD lead to a very rare neurodevelopmental disorder which is phenotypically pleiotropic grossly ranging from severe neonatal hypotonia, failure to thrive, multiple organ dysfunction, and early lethality to a similar but milder phenotype with better survival. Here, we report 5 patients from 3 unrelated Egyptian families in whom 4 patients showed the severe end of the spectrum displaying neonatal respiratory distress, hypotonia and chronic diarrhea while one patient presented with the mild form displaying moderate intellectual disability and myopathy. In addition, we observed distal arthrogryposis and nonspecific structural brain anomalies in all our patients. Interestingly, cerebellar and brainstem hypoplasia were noted in one patient. Whole exome sequencing identified three novel homozygous variants in the MADD gene: two likely pathogenic [c.4321delC p.(Gln1441ArgfsTer46) and c.2620 C > T p.(Arg874Ter)] and one variant of uncertain significance (c.4307 G > A, p.Arg1436Gln). The variants segregated with the disease in all available family members. Our findings confirm that arthrogryposis, genital, cardiac and structural brain anomalies are manifestations of MADD which expand the spectrum of MADD-related neurodevelopmental disorder. Moreover, they further highlight the convergence of MADD variants on different organ systems leading to complex phenotypes.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
