{"title":"不同铁(II)-三吡啶衍生物光谱行为的 DFT 研究及其在 DSSC 中的应用。","authors":"Evangelia Athanasopoulos, Jeanet Conradie","doi":"10.1016/j.jmgm.2024.108753","DOIUrl":null,"url":null,"abstract":"<div><p>Through a comprehensive computational analysis utilizing Density Functional Theory (DFT), we clarify the electronic structure and spectroscopic properties of modified iron(II)-terpyridine derivatives, with the aim of enhancing the efficiency of Dye-Sensitized Solar Cells (DSSCs). We optimized a series of nineteen iron(II)-terpyridine derivatives and related compounds in acetonitrile (MeCN) as the solvent using TDDFT, evaluating their potential as dyes for DSSCs. From the conducted computations on the optimized geometries of the nineteen [Fe(L<sup>n</sup>)<sub>2</sub>]<sup>2+</sup> complexes, containing substituted terpyridine and related ligands L<sup>1</sup>-L<sup>19</sup>, we determined the wavelengths (<em>λ</em> in nm), transition energy (<em>E</em> in eV), oscillator strength (<em>f</em>), type of transitions, excited state lifetime (<em>τ</em>), light harvesting efficiency (LHE), frontier orbital character and their energies (<em>E</em><sub>LUMO</sub>/<em>E</em><sub>HOMO</sub>), natural transition orbitals (NTOs), injection driving force of a dye (Δ<em>G</em><sub>inject</sub>), and regeneration driving force of a dye (Δ<em>G</em><sub>regenerate</sub>). Results show that the theoretically calculated values for assessing dye efficiency in a DSSC correlate with available experimental values. The UV–visible spectra of [Fe(L<sup>n</sup>)<sub>2</sub>]<sup>2+</sup> exhibited a peak above 500 nm (<em>λ</em><sub>max</sub>) in the visible region, attributed to the ligand-to-metal charge transfer band (LMCT) in literature, and a significant absorbance peak at approximately 300 nm (<em>λ</em><sub>A,max</sub>) in the UV region. The M06-D3/CEP-121G method replicated all reported <em>λ</em><sub>max</sub> and <em>λ</em><sub>A,max</sub> values with a mean absolute deviation (MAD) of 21 and 18 nm, respectively. Our findings underscore the connections between electronic modifications and absorption spectra, emphasizing their impact on the light-harvesting capabilities and overall performance of DSSCs. This research contributes to the advancement of fundamental principles governing the design and optimization of novel photovoltaic materials, facilitating the development of more efficient and sustainable solar energy technologies.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"129 ","pages":"Article 108753"},"PeriodicalIF":3.1000,"publicationDate":"2024-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S1093326324000536/pdfft?md5=e57be4ef77e0e23c32eb385ec4657676&pid=1-s2.0-S1093326324000536-main.pdf","citationCount":"0","resultStr":"{\"title\":\"DFT study of the spectroscopic behaviour of different iron(II)-terpyridine derivatives with application in DSSCs\",\"authors\":\"Evangelia Athanasopoulos, Jeanet Conradie\",\"doi\":\"10.1016/j.jmgm.2024.108753\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Through a comprehensive computational analysis utilizing Density Functional Theory (DFT), we clarify the electronic structure and spectroscopic properties of modified iron(II)-terpyridine derivatives, with the aim of enhancing the efficiency of Dye-Sensitized Solar Cells (DSSCs). We optimized a series of nineteen iron(II)-terpyridine derivatives and related compounds in acetonitrile (MeCN) as the solvent using TDDFT, evaluating their potential as dyes for DSSCs. From the conducted computations on the optimized geometries of the nineteen [Fe(L<sup>n</sup>)<sub>2</sub>]<sup>2+</sup> complexes, containing substituted terpyridine and related ligands L<sup>1</sup>-L<sup>19</sup>, we determined the wavelengths (<em>λ</em> in nm), transition energy (<em>E</em> in eV), oscillator strength (<em>f</em>), type of transitions, excited state lifetime (<em>τ</em>), light harvesting efficiency (LHE), frontier orbital character and their energies (<em>E</em><sub>LUMO</sub>/<em>E</em><sub>HOMO</sub>), natural transition orbitals (NTOs), injection driving force of a dye (Δ<em>G</em><sub>inject</sub>), and regeneration driving force of a dye (Δ<em>G</em><sub>regenerate</sub>). Results show that the theoretically calculated values for assessing dye efficiency in a DSSC correlate with available experimental values. The UV–visible spectra of [Fe(L<sup>n</sup>)<sub>2</sub>]<sup>2+</sup> exhibited a peak above 500 nm (<em>λ</em><sub>max</sub>) in the visible region, attributed to the ligand-to-metal charge transfer band (LMCT) in literature, and a significant absorbance peak at approximately 300 nm (<em>λ</em><sub>A,max</sub>) in the UV region. The M06-D3/CEP-121G method replicated all reported <em>λ</em><sub>max</sub> and <em>λ</em><sub>A,max</sub> values with a mean absolute deviation (MAD) of 21 and 18 nm, respectively. Our findings underscore the connections between electronic modifications and absorption spectra, emphasizing their impact on the light-harvesting capabilities and overall performance of DSSCs. This research contributes to the advancement of fundamental principles governing the design and optimization of novel photovoltaic materials, facilitating the development of more efficient and sustainable solar energy technologies.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":\"129 \",\"pages\":\"Article 108753\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S1093326324000536/pdfft?md5=e57be4ef77e0e23c32eb385ec4657676&pid=1-s2.0-S1093326324000536-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324000536\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/3/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324000536","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/4 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
DFT study of the spectroscopic behaviour of different iron(II)-terpyridine derivatives with application in DSSCs
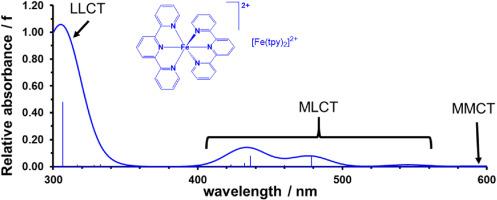
Through a comprehensive computational analysis utilizing Density Functional Theory (DFT), we clarify the electronic structure and spectroscopic properties of modified iron(II)-terpyridine derivatives, with the aim of enhancing the efficiency of Dye-Sensitized Solar Cells (DSSCs). We optimized a series of nineteen iron(II)-terpyridine derivatives and related compounds in acetonitrile (MeCN) as the solvent using TDDFT, evaluating their potential as dyes for DSSCs. From the conducted computations on the optimized geometries of the nineteen [Fe(Ln)2]2+ complexes, containing substituted terpyridine and related ligands L1-L19, we determined the wavelengths (λ in nm), transition energy (E in eV), oscillator strength (f), type of transitions, excited state lifetime (τ), light harvesting efficiency (LHE), frontier orbital character and their energies (ELUMO/EHOMO), natural transition orbitals (NTOs), injection driving force of a dye (ΔGinject), and regeneration driving force of a dye (ΔGregenerate). Results show that the theoretically calculated values for assessing dye efficiency in a DSSC correlate with available experimental values. The UV–visible spectra of [Fe(Ln)2]2+ exhibited a peak above 500 nm (λmax) in the visible region, attributed to the ligand-to-metal charge transfer band (LMCT) in literature, and a significant absorbance peak at approximately 300 nm (λA,max) in the UV region. The M06-D3/CEP-121G method replicated all reported λmax and λA,max values with a mean absolute deviation (MAD) of 21 and 18 nm, respectively. Our findings underscore the connections between electronic modifications and absorption spectra, emphasizing their impact on the light-harvesting capabilities and overall performance of DSSCs. This research contributes to the advancement of fundamental principles governing the design and optimization of novel photovoltaic materials, facilitating the development of more efficient and sustainable solar energy technologies.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
