Christopher J Record, Menelaos Pipis, Mariola Skorupinska, Julian Blake, Roy Poh, James M Polke, Kelly Eggleton, Tina Nanji, Stephan Zuchner, Andrea Cortese, Henry Houlden, Alexander M Rossor, Matilde Laura, Mary M Reilly
{"title":"全基因组测序提高了 Charcot-Marie-Tooth 病的诊断率。","authors":"Christopher J Record, Menelaos Pipis, Mariola Skorupinska, Julian Blake, Roy Poh, James M Polke, Kelly Eggleton, Tina Nanji, Stephan Zuchner, Andrea Cortese, Henry Houlden, Alexander M Rossor, Matilde Laura, Mary M Reilly","doi":"10.1093/brain/awae064","DOIUrl":null,"url":null,"abstract":"<p><p>Charcot-Marie-Tooth disease (CMT) is one of the most common and genetically heterogeneous inherited neurological diseases, with more than 130 disease-causing genes. Whole genome sequencing (WGS) has improved diagnosis across genetic diseases, but the diagnostic impact in CMT is yet to be fully reported. We present the diagnostic results from a single specialist inherited neuropathy centre, including the impact of WGS diagnostic testing. Patients were assessed at our specialist inherited neuropathy centre from 2009 to 2023. Genetic testing was performed using single gene testing, next-generation sequencing targeted panels, research whole exome sequencing and WGS and, latterly, WGS through the UK National Health Service. Variants were assessed using the American College of Medical Genetics and Genomics and Association for Clinical Genomic Science criteria. Excluding patients with hereditary ATTR amyloidosis, 1515 patients with a clinical diagnosis of CMT and related disorders were recruited. In summary, 621 patients had CMT1 (41.0%), 294 CMT2 (19.4%), 205 intermediate CMT (CMTi, 13.5%), 139 hereditary motor neuropathy (HMN, 9.2%), 93 hereditary sensory neuropathy (HSN, 6.1%), 38 sensory ataxic neuropathy (2.5%), 72 hereditary neuropathy with liability to pressure palsies (HNPP, 4.8%) and 53 'complex' neuropathy (3.5%). Overall, a genetic diagnosis was reached in 76.9% (1165/1515). A diagnosis was most likely in CMT1 (96.8%, 601/621), followed by CMTi (81.0%, 166/205) and then HSN (69.9%, 65/93). Diagnostic rates remained less than 50% in CMT2, HMN and complex neuropathies. The most common genetic diagnosis was PMP22 duplication (CMT1A; 505/1165, 43.3%), then GJB1 (CMTX1; 151/1165, 13.0%), PMP22 deletion (HNPP; 72/1165, 6.2%) and MFN2 (CMT2A; 46/1165, 3.9%). We recruited 233 cases to the UK 100 000 Genomes Project (100KGP), of which 74 (31.8%) achieved a diagnosis; 28 had been otherwise diagnosed since recruitment, leaving a true diagnostic rate of WGS through the 100KGP of 19.7% (46/233). However, almost half of the solved cases (35/74) received a negative report from the study, and the diagnosis was made through our research access to the WGS data. The overall diagnostic uplift of WGS for the entire cohort was 3.5%. Our diagnostic rate is the highest reported from a single centre and has benefitted from the use of WGS, particularly access to the raw data. However, almost one-quarter of all cases remain unsolved, and a new reference genome and novel technologies will be important to narrow the 'diagnostic gap'.</p>","PeriodicalId":9063,"journal":{"name":"Brain","volume":" ","pages":"3144-3156"},"PeriodicalIF":11.7000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11370804/pdf/","citationCount":"0","resultStr":"{\"title\":\"Whole genome sequencing increases the diagnostic rate in Charcot-Marie-Tooth disease.\",\"authors\":\"Christopher J Record, Menelaos Pipis, Mariola Skorupinska, Julian Blake, Roy Poh, James M Polke, Kelly Eggleton, Tina Nanji, Stephan Zuchner, Andrea Cortese, Henry Houlden, Alexander M Rossor, Matilde Laura, Mary M Reilly\",\"doi\":\"10.1093/brain/awae064\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Charcot-Marie-Tooth disease (CMT) is one of the most common and genetically heterogeneous inherited neurological diseases, with more than 130 disease-causing genes. Whole genome sequencing (WGS) has improved diagnosis across genetic diseases, but the diagnostic impact in CMT is yet to be fully reported. We present the diagnostic results from a single specialist inherited neuropathy centre, including the impact of WGS diagnostic testing. Patients were assessed at our specialist inherited neuropathy centre from 2009 to 2023. Genetic testing was performed using single gene testing, next-generation sequencing targeted panels, research whole exome sequencing and WGS and, latterly, WGS through the UK National Health Service. Variants were assessed using the American College of Medical Genetics and Genomics and Association for Clinical Genomic Science criteria. Excluding patients with hereditary ATTR amyloidosis, 1515 patients with a clinical diagnosis of CMT and related disorders were recruited. In summary, 621 patients had CMT1 (41.0%), 294 CMT2 (19.4%), 205 intermediate CMT (CMTi, 13.5%), 139 hereditary motor neuropathy (HMN, 9.2%), 93 hereditary sensory neuropathy (HSN, 6.1%), 38 sensory ataxic neuropathy (2.5%), 72 hereditary neuropathy with liability to pressure palsies (HNPP, 4.8%) and 53 'complex' neuropathy (3.5%). Overall, a genetic diagnosis was reached in 76.9% (1165/1515). A diagnosis was most likely in CMT1 (96.8%, 601/621), followed by CMTi (81.0%, 166/205) and then HSN (69.9%, 65/93). Diagnostic rates remained less than 50% in CMT2, HMN and complex neuropathies. The most common genetic diagnosis was PMP22 duplication (CMT1A; 505/1165, 43.3%), then GJB1 (CMTX1; 151/1165, 13.0%), PMP22 deletion (HNPP; 72/1165, 6.2%) and MFN2 (CMT2A; 46/1165, 3.9%). We recruited 233 cases to the UK 100 000 Genomes Project (100KGP), of which 74 (31.8%) achieved a diagnosis; 28 had been otherwise diagnosed since recruitment, leaving a true diagnostic rate of WGS through the 100KGP of 19.7% (46/233). However, almost half of the solved cases (35/74) received a negative report from the study, and the diagnosis was made through our research access to the WGS data. The overall diagnostic uplift of WGS for the entire cohort was 3.5%. Our diagnostic rate is the highest reported from a single centre and has benefitted from the use of WGS, particularly access to the raw data. However, almost one-quarter of all cases remain unsolved, and a new reference genome and novel technologies will be important to narrow the 'diagnostic gap'.</p>\",\"PeriodicalId\":9063,\"journal\":{\"name\":\"Brain\",\"volume\":\" \",\"pages\":\"3144-3156\"},\"PeriodicalIF\":11.7000,\"publicationDate\":\"2024-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11370804/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Brain\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1093/brain/awae064\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/brain/awae064","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Whole genome sequencing increases the diagnostic rate in Charcot-Marie-Tooth disease.
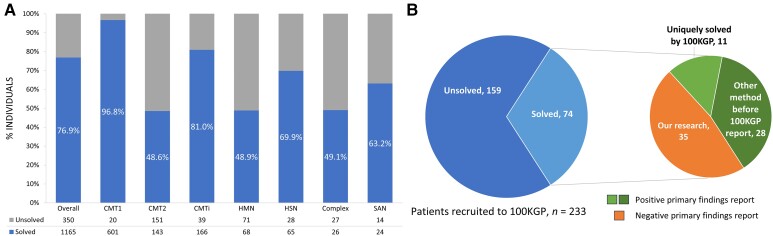
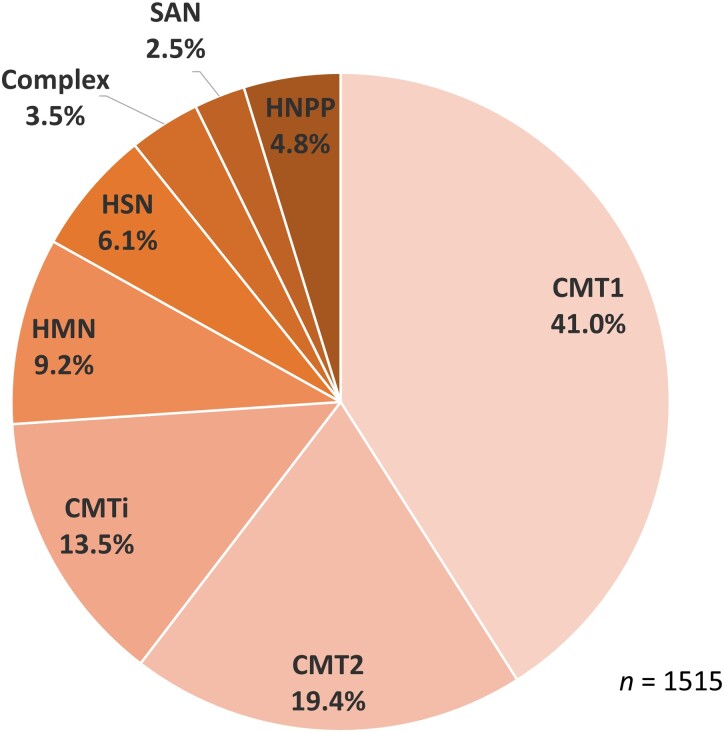
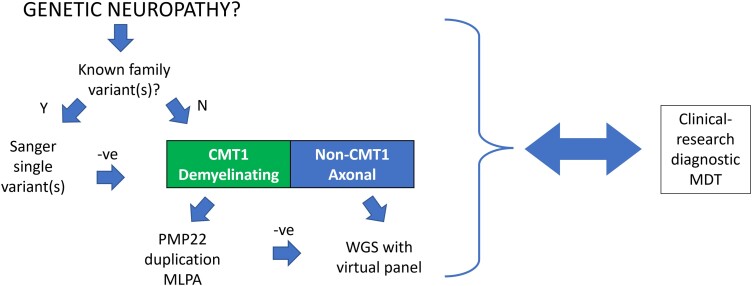
Charcot-Marie-Tooth disease (CMT) is one of the most common and genetically heterogeneous inherited neurological diseases, with more than 130 disease-causing genes. Whole genome sequencing (WGS) has improved diagnosis across genetic diseases, but the diagnostic impact in CMT is yet to be fully reported. We present the diagnostic results from a single specialist inherited neuropathy centre, including the impact of WGS diagnostic testing. Patients were assessed at our specialist inherited neuropathy centre from 2009 to 2023. Genetic testing was performed using single gene testing, next-generation sequencing targeted panels, research whole exome sequencing and WGS and, latterly, WGS through the UK National Health Service. Variants were assessed using the American College of Medical Genetics and Genomics and Association for Clinical Genomic Science criteria. Excluding patients with hereditary ATTR amyloidosis, 1515 patients with a clinical diagnosis of CMT and related disorders were recruited. In summary, 621 patients had CMT1 (41.0%), 294 CMT2 (19.4%), 205 intermediate CMT (CMTi, 13.5%), 139 hereditary motor neuropathy (HMN, 9.2%), 93 hereditary sensory neuropathy (HSN, 6.1%), 38 sensory ataxic neuropathy (2.5%), 72 hereditary neuropathy with liability to pressure palsies (HNPP, 4.8%) and 53 'complex' neuropathy (3.5%). Overall, a genetic diagnosis was reached in 76.9% (1165/1515). A diagnosis was most likely in CMT1 (96.8%, 601/621), followed by CMTi (81.0%, 166/205) and then HSN (69.9%, 65/93). Diagnostic rates remained less than 50% in CMT2, HMN and complex neuropathies. The most common genetic diagnosis was PMP22 duplication (CMT1A; 505/1165, 43.3%), then GJB1 (CMTX1; 151/1165, 13.0%), PMP22 deletion (HNPP; 72/1165, 6.2%) and MFN2 (CMT2A; 46/1165, 3.9%). We recruited 233 cases to the UK 100 000 Genomes Project (100KGP), of which 74 (31.8%) achieved a diagnosis; 28 had been otherwise diagnosed since recruitment, leaving a true diagnostic rate of WGS through the 100KGP of 19.7% (46/233). However, almost half of the solved cases (35/74) received a negative report from the study, and the diagnosis was made through our research access to the WGS data. The overall diagnostic uplift of WGS for the entire cohort was 3.5%. Our diagnostic rate is the highest reported from a single centre and has benefitted from the use of WGS, particularly access to the raw data. However, almost one-quarter of all cases remain unsolved, and a new reference genome and novel technologies will be important to narrow the 'diagnostic gap'.
期刊介绍:
Brain, a journal focused on clinical neurology and translational neuroscience, has been publishing landmark papers since 1878. The journal aims to expand its scope by including studies that shed light on disease mechanisms and conducting innovative clinical trials for brain disorders. With a wide range of topics covered, the Editorial Board represents the international readership and diverse coverage of the journal. Accepted articles are promptly posted online, typically within a few weeks of acceptance. As of 2022, Brain holds an impressive impact factor of 14.5, according to the Journal Citation Reports.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
