Natalia Martínez Córdoba, Isabella Lince-Rivera, Jorge Luis Ramón Gómez, Guido Rubboli, Sebastián Ortiz De la Rosa
{"title":"ATP1A2相关癫痫性脑病和运动障碍:三名新患者的临床特征","authors":"Natalia Martínez Córdoba, Isabella Lince-Rivera, Jorge Luis Ramón Gómez, Guido Rubboli, Sebastián Ortiz De la Rosa","doi":"10.1002/epd2.20220","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Objective</h3>\n \n <p>Variants in the <i>ATP1A2</i> gene exhibit a wide clinical spectrum, ranging from familial hemiplegic migraine to childhood epilepsies and early infantile developmental epileptic encephalopathy (EIDEE) with movement disorders. This study aims to describe the epileptology of three unpublished cases and summarize epilepsy features of the other 17 published cases with <i>ATP1A2</i> variants and EIDEE.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>Medical records of three novel patients with pathogenic <i>ATP1A2</i> variants were retrospectively reviewed. Additionally, the PUBMED, EMBASE, and Cochrane databases were searched until December 2023 for articles on EIDEE with ATP1A2 variants, without language or publication year restrictions.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Three female patients, aged 6 months–10 years, were investigated. Epilepsy onset occurred between 5 days and 2 years, accompanied by severe developmental delay, intellectual disability, drug-resistant epilepsy, severe movement disorder, and recurrent status epilepticus. All individuals had pathogenic variants of the <i>ATP1A2</i> gene (<i>ATP1A2</i> c.720_721del (p.Ile240MetfsTer9), <i>ATP1A2c.3022C > T (p.Arg1008Trp)</i>, <i>ATP1A2 c.1096G > T (p.Gly366Cys)</i>, according to ACMG criteria. Memantine was p) rescribed to three patients, one with a reduction in ictal frequency, one with improvement in gait pattern, coordination, and attention span, and another one in alertness without significant side effects.</p>\n </section>\n \n <section>\n \n <h3> Significance</h3>\n \n <p>This study reinforces the association between <i>ATP1A2</i> variants and a severe phenotype. All patients had de novo variants, focal motor seizures with impaired awareness as the primary type of seizure; of the 11 EEGs recorded, 10 presented a slow background rhythm, 7 multifocal interictal epileptiform discharges (IED), predominantly temporal IEDs, followed by frontal IED, as well as ten ictal recordings, which showed ictal onset from the same regions mentioned above. Treatment with antiseizure medication was generally ineffective, but memantine showed moderate improvement. Prospective studies are needed to enlarge the phenotype and assess the efficacy of NMDA receptor antagonist therapies in reducing seizure frequency and improving quality of life.</p>\n </section>\n </div>","PeriodicalId":50508,"journal":{"name":"Epileptic Disorders","volume":"26 3","pages":"332-340"},"PeriodicalIF":2.7000,"publicationDate":"2024-03-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/epd2.20220","citationCount":"0","resultStr":"{\"title\":\"ATP1A2-related epileptic encephalopathy and movement disorder: Clinical features of three novel patients\",\"authors\":\"Natalia Martínez Córdoba, Isabella Lince-Rivera, Jorge Luis Ramón Gómez, Guido Rubboli, Sebastián Ortiz De la Rosa\",\"doi\":\"10.1002/epd2.20220\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Objective</h3>\\n \\n <p>Variants in the <i>ATP1A2</i> gene exhibit a wide clinical spectrum, ranging from familial hemiplegic migraine to childhood epilepsies and early infantile developmental epileptic encephalopathy (EIDEE) with movement disorders. This study aims to describe the epileptology of three unpublished cases and summarize epilepsy features of the other 17 published cases with <i>ATP1A2</i> variants and EIDEE.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>Medical records of three novel patients with pathogenic <i>ATP1A2</i> variants were retrospectively reviewed. Additionally, the PUBMED, EMBASE, and Cochrane databases were searched until December 2023 for articles on EIDEE with ATP1A2 variants, without language or publication year restrictions.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Three female patients, aged 6 months–10 years, were investigated. Epilepsy onset occurred between 5 days and 2 years, accompanied by severe developmental delay, intellectual disability, drug-resistant epilepsy, severe movement disorder, and recurrent status epilepticus. All individuals had pathogenic variants of the <i>ATP1A2</i> gene (<i>ATP1A2</i> c.720_721del (p.Ile240MetfsTer9), <i>ATP1A2c.3022C > T (p.Arg1008Trp)</i>, <i>ATP1A2 c.1096G > T (p.Gly366Cys)</i>, according to ACMG criteria. Memantine was p) rescribed to three patients, one with a reduction in ictal frequency, one with improvement in gait pattern, coordination, and attention span, and another one in alertness without significant side effects.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Significance</h3>\\n \\n <p>This study reinforces the association between <i>ATP1A2</i> variants and a severe phenotype. All patients had de novo variants, focal motor seizures with impaired awareness as the primary type of seizure; of the 11 EEGs recorded, 10 presented a slow background rhythm, 7 multifocal interictal epileptiform discharges (IED), predominantly temporal IEDs, followed by frontal IED, as well as ten ictal recordings, which showed ictal onset from the same regions mentioned above. Treatment with antiseizure medication was generally ineffective, but memantine showed moderate improvement. Prospective studies are needed to enlarge the phenotype and assess the efficacy of NMDA receptor antagonist therapies in reducing seizure frequency and improving quality of life.</p>\\n </section>\\n </div>\",\"PeriodicalId\":50508,\"journal\":{\"name\":\"Epileptic Disorders\",\"volume\":\"26 3\",\"pages\":\"332-340\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-03-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/epd2.20220\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Epileptic Disorders\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/epd2.20220\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epileptic Disorders","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/epd2.20220","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
ATP1A2-related epileptic encephalopathy and movement disorder: Clinical features of three novel patients
Objective
Variants in the ATP1A2 gene exhibit a wide clinical spectrum, ranging from familial hemiplegic migraine to childhood epilepsies and early infantile developmental epileptic encephalopathy (EIDEE) with movement disorders. This study aims to describe the epileptology of three unpublished cases and summarize epilepsy features of the other 17 published cases with ATP1A2 variants and EIDEE.
Methods
Medical records of three novel patients with pathogenic ATP1A2 variants were retrospectively reviewed. Additionally, the PUBMED, EMBASE, and Cochrane databases were searched until December 2023 for articles on EIDEE with ATP1A2 variants, without language or publication year restrictions.
Results
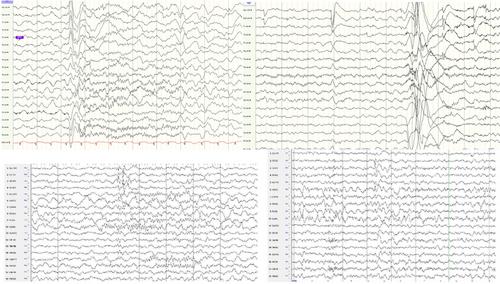
Three female patients, aged 6 months–10 years, were investigated. Epilepsy onset occurred between 5 days and 2 years, accompanied by severe developmental delay, intellectual disability, drug-resistant epilepsy, severe movement disorder, and recurrent status epilepticus. All individuals had pathogenic variants of the ATP1A2 gene (ATP1A2 c.720_721del (p.Ile240MetfsTer9), ATP1A2c.3022C > T (p.Arg1008Trp), ATP1A2 c.1096G > T (p.Gly366Cys), according to ACMG criteria. Memantine was p) rescribed to three patients, one with a reduction in ictal frequency, one with improvement in gait pattern, coordination, and attention span, and another one in alertness without significant side effects.
Significance
This study reinforces the association between ATP1A2 variants and a severe phenotype. All patients had de novo variants, focal motor seizures with impaired awareness as the primary type of seizure; of the 11 EEGs recorded, 10 presented a slow background rhythm, 7 multifocal interictal epileptiform discharges (IED), predominantly temporal IEDs, followed by frontal IED, as well as ten ictal recordings, which showed ictal onset from the same regions mentioned above. Treatment with antiseizure medication was generally ineffective, but memantine showed moderate improvement. Prospective studies are needed to enlarge the phenotype and assess the efficacy of NMDA receptor antagonist therapies in reducing seizure frequency and improving quality of life.
期刊介绍:
Epileptic Disorders is the leading forum where all experts and medical studentswho wish to improve their understanding of epilepsy and related disorders can share practical experiences surrounding diagnosis and care, natural history, and management of seizures.
Epileptic Disorders is the official E-journal of the International League Against Epilepsy for educational communication. As the journal celebrates its 20th anniversary, it will now be available only as an online version. Its mission is to create educational links between epileptologists and other health professionals in clinical practice and scientists or physicians in research-based institutions. This change is accompanied by an increase in the number of issues per year, from 4 to 6, to ensure regular diffusion of recently published material (high quality Review and Seminar in Epileptology papers; Original Research articles or Case reports of educational value; MultiMedia Teaching Material), to serve the global medical community that cares for those affected by epilepsy.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
