Charlotte Herbst, Viktoria Bothe, Meret Wegler, Susanne Axer-Schaefer, Séverine Audebert-Bellanger, Jozef Gecz, Benjamin Cogne, Hagit Baris Feldman, Anselm H C Horn, Anna C E Hurst, Melissa A Kelly, Michael C Kruer, Alina Kurolap, Annie Laquerriere, Megan Li, Paul R Mark, Markus Morawski, Mathilde Nizon, Tomi Pastinen, Tilman Polster, Pascale Saugier-Veber, Jang SeSong, Heinrich Sticht, Jens T Stieler, Isabelle Thifffault, Clare L van Eyk, Pascale Marcorelles, Myriam Vezain-Mouchard, Rami Abou Jamra, Henry Oppermann
{"title":"DOCK4 的杂合子功能缺失变体会导致神经发育迟缓和小头畸形。","authors":"Charlotte Herbst, Viktoria Bothe, Meret Wegler, Susanne Axer-Schaefer, Séverine Audebert-Bellanger, Jozef Gecz, Benjamin Cogne, Hagit Baris Feldman, Anselm H C Horn, Anna C E Hurst, Melissa A Kelly, Michael C Kruer, Alina Kurolap, Annie Laquerriere, Megan Li, Paul R Mark, Markus Morawski, Mathilde Nizon, Tomi Pastinen, Tilman Polster, Pascale Saugier-Veber, Jang SeSong, Heinrich Sticht, Jens T Stieler, Isabelle Thifffault, Clare L van Eyk, Pascale Marcorelles, Myriam Vezain-Mouchard, Rami Abou Jamra, Henry Oppermann","doi":"10.1007/s00439-024-02655-4","DOIUrl":null,"url":null,"abstract":"<p><p>Neurons form the basic anatomical and functional structure of the nervous system, and defects in neuronal differentiation or formation of neurites are associated with various psychiatric and neurodevelopmental disorders. Dynamic changes in the cytoskeleton are essential for this process, which is, inter alia, controlled by the dedicator of cytokinesis 4 (DOCK4) through the activation of RAC1. Here, we clinically describe 7 individuals (6 males and one female) with variants in DOCK4 and overlapping phenotype of mild to severe global developmental delay. Additional symptoms include coordination or gait abnormalities, microcephaly, nonspecific brain malformations, hypotonia and seizures. Four individuals carry missense variants (three of them detected de novo) and three individuals carry null variants (two of them maternally inherited). Molecular modeling of the heterozygous missense variants suggests that the majority of them affect the globular structure of DOCK4. In vitro functional expression studies in transfected Neuro-2A cells showed that all missense variants impaired neurite outgrowth. Furthermore, Dock4 knockout Neuro-2A cells also exhibited defects in promoting neurite outgrowth. Our results, including clinical, molecular and functional data, suggest that loss-of-function variants in DOCK4 probable cause a variable spectrum of a novel neurodevelopmental disorder with microcephaly.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"455-469"},"PeriodicalIF":3.6000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11043173/pdf/","citationCount":"0","resultStr":"{\"title\":\"Heterozygous loss-of-function variants in DOCK4 cause neurodevelopmental delay and microcephaly.\",\"authors\":\"Charlotte Herbst, Viktoria Bothe, Meret Wegler, Susanne Axer-Schaefer, Séverine Audebert-Bellanger, Jozef Gecz, Benjamin Cogne, Hagit Baris Feldman, Anselm H C Horn, Anna C E Hurst, Melissa A Kelly, Michael C Kruer, Alina Kurolap, Annie Laquerriere, Megan Li, Paul R Mark, Markus Morawski, Mathilde Nizon, Tomi Pastinen, Tilman Polster, Pascale Saugier-Veber, Jang SeSong, Heinrich Sticht, Jens T Stieler, Isabelle Thifffault, Clare L van Eyk, Pascale Marcorelles, Myriam Vezain-Mouchard, Rami Abou Jamra, Henry Oppermann\",\"doi\":\"10.1007/s00439-024-02655-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Neurons form the basic anatomical and functional structure of the nervous system, and defects in neuronal differentiation or formation of neurites are associated with various psychiatric and neurodevelopmental disorders. Dynamic changes in the cytoskeleton are essential for this process, which is, inter alia, controlled by the dedicator of cytokinesis 4 (DOCK4) through the activation of RAC1. Here, we clinically describe 7 individuals (6 males and one female) with variants in DOCK4 and overlapping phenotype of mild to severe global developmental delay. Additional symptoms include coordination or gait abnormalities, microcephaly, nonspecific brain malformations, hypotonia and seizures. Four individuals carry missense variants (three of them detected de novo) and three individuals carry null variants (two of them maternally inherited). Molecular modeling of the heterozygous missense variants suggests that the majority of them affect the globular structure of DOCK4. In vitro functional expression studies in transfected Neuro-2A cells showed that all missense variants impaired neurite outgrowth. Furthermore, Dock4 knockout Neuro-2A cells also exhibited defects in promoting neurite outgrowth. Our results, including clinical, molecular and functional data, suggest that loss-of-function variants in DOCK4 probable cause a variable spectrum of a novel neurodevelopmental disorder with microcephaly.</p>\",\"PeriodicalId\":13175,\"journal\":{\"name\":\"Human Genetics\",\"volume\":\" \",\"pages\":\"455-469\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11043173/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s00439-024-02655-4\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/3/25 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02655-4","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/25 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Heterozygous loss-of-function variants in DOCK4 cause neurodevelopmental delay and microcephaly.
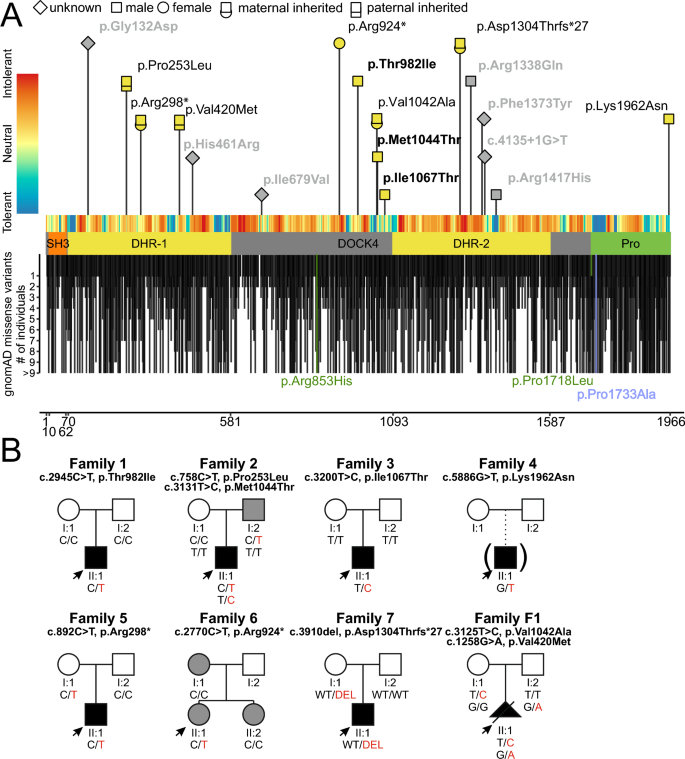
Neurons form the basic anatomical and functional structure of the nervous system, and defects in neuronal differentiation or formation of neurites are associated with various psychiatric and neurodevelopmental disorders. Dynamic changes in the cytoskeleton are essential for this process, which is, inter alia, controlled by the dedicator of cytokinesis 4 (DOCK4) through the activation of RAC1. Here, we clinically describe 7 individuals (6 males and one female) with variants in DOCK4 and overlapping phenotype of mild to severe global developmental delay. Additional symptoms include coordination or gait abnormalities, microcephaly, nonspecific brain malformations, hypotonia and seizures. Four individuals carry missense variants (three of them detected de novo) and three individuals carry null variants (two of them maternally inherited). Molecular modeling of the heterozygous missense variants suggests that the majority of them affect the globular structure of DOCK4. In vitro functional expression studies in transfected Neuro-2A cells showed that all missense variants impaired neurite outgrowth. Furthermore, Dock4 knockout Neuro-2A cells also exhibited defects in promoting neurite outgrowth. Our results, including clinical, molecular and functional data, suggest that loss-of-function variants in DOCK4 probable cause a variable spectrum of a novel neurodevelopmental disorder with microcephaly.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
