Ming-Der Lin , Chia-Hsien Chuang , Chih-Hsin Kao , Shu-Hwa Chen , Szu-Chieh Wang , Ping-Heng Hsieh , Guan-Yu Chen , Chun-Chia Mao , Jeng-Yi Li , Mei-Yeh Jade Lu , Chung-Yen Lin
{"title":"解码吸血蠓 Forcipomyia taiwana(双翅目:Ceratopogonidae)的基因组:对气味受体扩展的启示","authors":"Ming-Der Lin , Chia-Hsien Chuang , Chih-Hsin Kao , Shu-Hwa Chen , Szu-Chieh Wang , Ping-Heng Hsieh , Guan-Yu Chen , Chun-Chia Mao , Jeng-Yi Li , Mei-Yeh Jade Lu , Chung-Yen Lin","doi":"10.1016/j.ibmb.2024.104115","DOIUrl":null,"url":null,"abstract":"<div><p>Biting midges, notably those within the Ceratopogonidae family, have long been recognized for their epidemiological significance, both as nuisances and vectors for disease transmission in vertebrates. Despite their impact, genomic insights into these insects, particularly beyond the <em>Culicoides</em> genus, remain limited. In this study, we assembled the <em>Forcipomyia taiwana</em> (Shiraki) genome, comprising 113 scaffolds covering 130.4 Mbps—with the longest scaffold reaching 7.6 Mbps and an N50 value of 2.6 Mbps—marking a pivotal advancement in understanding the genetic architecture of ceratopogonid biting midges. Phylogenomic analyses reveal a shared ancestry between <em>F. taiwana</em> and <em>Culicoides sonorensis</em> Wirth & Jones, dating back approximately 124 million years, and highlight a dynamic history of gene family expansions and contractions within the Ceratopogonidae family. Notably, a substantial expansion of the <em>odorant receptor</em> (<em>OR</em>) gene family was observed, which is crucial for the chemosensory capabilities that govern biting midges' interactions with their environment, including host seeking and oviposition behaviors. The distribution of <em>OR</em> genes across the <em>F. taiwana</em> genome displays notable clusters on scaffolds, indicating localized tandem gene duplication events. Additionally, several collinear regions were identified, hinting at segmental duplications, inversions, and translocations, contributing to the olfactory system's evolutionary complexity. Among the 156 ORs identified in <em>F. taiwana</em>, 134 are biting midge-specific ORs, distributed across three distinct clades, each exhibiting unique motif features that distinguish them from the others. Through weighted gene co-expression network analysis, we correlated distinct gene modules with sex and reproductive status, laying the groundwork for future investigations into the interplay between gene expression and adaptive behaviors in <em>F. taiwana</em>. In conclusion, our study not only highlights the unique olfactory repertoire of ceratopogonid biting midges but also sets the stage for future studies into the genetic underpinnings of their unique biological traits and ecological strategies.</p></div>","PeriodicalId":330,"journal":{"name":"Insect Biochemistry and Molecular Biology","volume":"168 ","pages":"Article 104115"},"PeriodicalIF":4.3000,"publicationDate":"2024-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Decoding the genome of bloodsucking midge Forcipomyia taiwana (Diptera: Ceratopogonidae): Insights into odorant receptor expansion\",\"authors\":\"Ming-Der Lin , Chia-Hsien Chuang , Chih-Hsin Kao , Shu-Hwa Chen , Szu-Chieh Wang , Ping-Heng Hsieh , Guan-Yu Chen , Chun-Chia Mao , Jeng-Yi Li , Mei-Yeh Jade Lu , Chung-Yen Lin\",\"doi\":\"10.1016/j.ibmb.2024.104115\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Biting midges, notably those within the Ceratopogonidae family, have long been recognized for their epidemiological significance, both as nuisances and vectors for disease transmission in vertebrates. Despite their impact, genomic insights into these insects, particularly beyond the <em>Culicoides</em> genus, remain limited. In this study, we assembled the <em>Forcipomyia taiwana</em> (Shiraki) genome, comprising 113 scaffolds covering 130.4 Mbps—with the longest scaffold reaching 7.6 Mbps and an N50 value of 2.6 Mbps—marking a pivotal advancement in understanding the genetic architecture of ceratopogonid biting midges. Phylogenomic analyses reveal a shared ancestry between <em>F. taiwana</em> and <em>Culicoides sonorensis</em> Wirth & Jones, dating back approximately 124 million years, and highlight a dynamic history of gene family expansions and contractions within the Ceratopogonidae family. Notably, a substantial expansion of the <em>odorant receptor</em> (<em>OR</em>) gene family was observed, which is crucial for the chemosensory capabilities that govern biting midges' interactions with their environment, including host seeking and oviposition behaviors. The distribution of <em>OR</em> genes across the <em>F. taiwana</em> genome displays notable clusters on scaffolds, indicating localized tandem gene duplication events. Additionally, several collinear regions were identified, hinting at segmental duplications, inversions, and translocations, contributing to the olfactory system's evolutionary complexity. Among the 156 ORs identified in <em>F. taiwana</em>, 134 are biting midge-specific ORs, distributed across three distinct clades, each exhibiting unique motif features that distinguish them from the others. Through weighted gene co-expression network analysis, we correlated distinct gene modules with sex and reproductive status, laying the groundwork for future investigations into the interplay between gene expression and adaptive behaviors in <em>F. taiwana</em>. In conclusion, our study not only highlights the unique olfactory repertoire of ceratopogonid biting midges but also sets the stage for future studies into the genetic underpinnings of their unique biological traits and ecological strategies.</p></div>\",\"PeriodicalId\":330,\"journal\":{\"name\":\"Insect Biochemistry and Molecular Biology\",\"volume\":\"168 \",\"pages\":\"Article 104115\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2024-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Insect Biochemistry and Molecular Biology\",\"FirstCategoryId\":\"97\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0965174824000468\",\"RegionNum\":2,\"RegionCategory\":\"农林科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/4/1 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Insect Biochemistry and Molecular Biology","FirstCategoryId":"97","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0965174824000468","RegionNum":2,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/4/1 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
咬蠓,尤其是咬蠓科中的咬蠓,长期以来一直被认为具有重要的流行病学意义,既是害虫,也是脊椎动物疾病传播的媒介。尽管这些昆虫具有重要影响,但对它们的基因组学研究仍然有限,尤其是在Culicoides属以外的昆虫。在这项研究中,我们组装了太瓦喙蠓(Forcipomyia taiwana,Shiraki)的基因组,包括 113 个脚架,覆盖 130.4 Mbps,其中最长的脚架达到 7.6 Mbps,N50 值为 2.6 Mbps,这标志着我们在了解齿螨类咬蠓基因结构方面取得了关键性进展。系统发生组分析揭示了 F. taiwana 与 Culicoides sonorensis Wirth & Jones 之间的共同祖先,可追溯到约 1.24 亿年前,并突出了 Ceratopogonidae 家族内基因家族扩张和收缩的动态历史。值得注意的是,我们观察到气味受体(OR)基因家族的大幅扩展,这对于咬蠓与环境相互作用(包括寻找宿主和产卵行为)的化学感觉能力至关重要。OR 基因在 F. taiwana 基因组中的分布在支架上显示出明显的集群,表明发生了局部的串联基因重复事件。此外,研究还发现了几个共线区域,暗示着节段复制、倒位和易位,这也是嗅觉系统进化复杂性的原因之一。在 F. taiwana 中发现的 156 个 ORs 中,有 134 个是咬蠓特异性 ORs,分布在三个不同的支系中,每个支系都表现出区别于其他支系的独特图案特征。通过加权基因共表达网络分析,我们将不同的基因模块与性别和繁殖状态相关联,为今后研究太瓦纳蝇基因表达与适应行为之间的相互作用奠定了基础。总之,我们的研究不仅突显了齿螨类咬蠓独特的嗅觉谱系,而且为今后研究其独特生物特征和生态策略的遗传基础奠定了基础。
Decoding the genome of bloodsucking midge Forcipomyia taiwana (Diptera: Ceratopogonidae): Insights into odorant receptor expansion
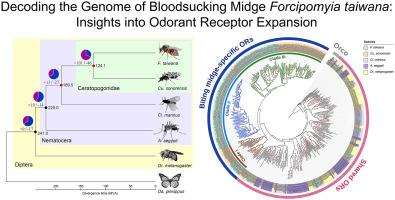
Biting midges, notably those within the Ceratopogonidae family, have long been recognized for their epidemiological significance, both as nuisances and vectors for disease transmission in vertebrates. Despite their impact, genomic insights into these insects, particularly beyond the Culicoides genus, remain limited. In this study, we assembled the Forcipomyia taiwana (Shiraki) genome, comprising 113 scaffolds covering 130.4 Mbps—with the longest scaffold reaching 7.6 Mbps and an N50 value of 2.6 Mbps—marking a pivotal advancement in understanding the genetic architecture of ceratopogonid biting midges. Phylogenomic analyses reveal a shared ancestry between F. taiwana and Culicoides sonorensis Wirth & Jones, dating back approximately 124 million years, and highlight a dynamic history of gene family expansions and contractions within the Ceratopogonidae family. Notably, a substantial expansion of the odorant receptor (OR) gene family was observed, which is crucial for the chemosensory capabilities that govern biting midges' interactions with their environment, including host seeking and oviposition behaviors. The distribution of OR genes across the F. taiwana genome displays notable clusters on scaffolds, indicating localized tandem gene duplication events. Additionally, several collinear regions were identified, hinting at segmental duplications, inversions, and translocations, contributing to the olfactory system's evolutionary complexity. Among the 156 ORs identified in F. taiwana, 134 are biting midge-specific ORs, distributed across three distinct clades, each exhibiting unique motif features that distinguish them from the others. Through weighted gene co-expression network analysis, we correlated distinct gene modules with sex and reproductive status, laying the groundwork for future investigations into the interplay between gene expression and adaptive behaviors in F. taiwana. In conclusion, our study not only highlights the unique olfactory repertoire of ceratopogonid biting midges but also sets the stage for future studies into the genetic underpinnings of their unique biological traits and ecological strategies.
期刊介绍:
This international journal publishes original contributions and mini-reviews in the fields of insect biochemistry and insect molecular biology. Main areas of interest are neurochemistry, hormone and pheromone biochemistry, enzymes and metabolism, hormone action and gene regulation, gene characterization and structure, pharmacology, immunology and cell and tissue culture. Papers on the biochemistry and molecular biology of other groups of arthropods are published if of general interest to the readership. Technique papers will be considered for publication if they significantly advance the field of insect biochemistry and molecular biology in the opinion of the Editors and Editorial Board.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
