{"title":"通过深度学习推进配体对接:虚拟筛选的挑战与前景","authors":"Xujun Zhang, Chao Shen, Haotian Zhang, Yu Kang, Chang-Yu Hsieh* and Tingjun Hou*, ","doi":"10.1021/acs.accounts.4c00093","DOIUrl":null,"url":null,"abstract":"<p >Molecular docking, also termed ligand docking (LD), is a pivotal element of structure-based virtual screening (SBVS) used to predict the binding conformations and affinities of protein–ligand complexes. Traditional LD methodologies rely on a search and scoring framework, utilizing heuristic algorithms to explore binding conformations and scoring functions to evaluate binding strengths. However, to meet the efficiency demands of SBVS, these algorithms and functions are often simplified, prioritizing speed over accuracy.</p><p >The emergence of deep learning (DL) has exerted a profound impact on diverse fields, ranging from natural language processing to computer vision and drug discovery. DeepMind’s AlphaFold2 has impressively exhibited its ability to accurately predict protein structures solely from amino acid sequences, highlighting the remarkable potential of DL in conformation prediction. This groundbreaking advancement circumvents the traditional search-scoring frameworks in LD, enhancing both accuracy and processing speed and thereby catalyzing a broader adoption of DL algorithms in binding pose prediction. Nevertheless, a consensus on certain aspects remains elusive.</p><p >In this Account, we delineate the current status of employing DL to augment LD within the VS paradigm, highlighting our contributions to this domain. Furthermore, we discuss the challenges and future prospects, drawing insights from our scholarly investigations. Initially, we present an overview of VS and LD, followed by an introduction to DL paradigms, which deviate significantly from traditional search-scoring frameworks. Subsequently, we delve into the challenges associated with the development of DL-based LD (DLLD), encompassing evaluation metrics, application scenarios, and physical plausibility of the predicted conformations. In the evaluation of LD algorithms, it is essential to recognize the multifaceted nature of the metrics. While the accuracy of binding pose prediction, often measured by the success rate, is a pivotal aspect, the scoring/screening power and computational speed of these algorithms are equally important given the pivotal role of LD tools in VS. Regarding application scenarios, early methods focused on blind docking, where the binding site is unknown. However, recent studies suggest a shift toward identifying binding sites rather than solely predicting binding poses within these models. In contrast, LD with a known pocket in VS has been shown to be more practical. Physical plausibility poses another significant challenge. Although DLLD models often achieve higher success rates compared to traditional methods, they may generate poses with implausible local structures, such as incorrect bond angles or lengths, which are disadvantageous for postprocessing tasks like visualization. Finally, we discuss the future perspectives for DLLD, emphasizing the need to improve generalization ability, strike a balance between speed and accuracy, account for protein conformation flexibility, and enhance physical plausibility. Additionally, we delve into the comparison between generative and regression algorithms in this context, exploring their respective strengths and potential.</p>","PeriodicalId":1,"journal":{"name":"Accounts of Chemical Research","volume":null,"pages":null},"PeriodicalIF":16.4000,"publicationDate":"2024-04-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Advancing Ligand Docking through Deep Learning: Challenges and Prospects in Virtual Screening\",\"authors\":\"Xujun Zhang, Chao Shen, Haotian Zhang, Yu Kang, Chang-Yu Hsieh* and Tingjun Hou*, \",\"doi\":\"10.1021/acs.accounts.4c00093\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Molecular docking, also termed ligand docking (LD), is a pivotal element of structure-based virtual screening (SBVS) used to predict the binding conformations and affinities of protein–ligand complexes. Traditional LD methodologies rely on a search and scoring framework, utilizing heuristic algorithms to explore binding conformations and scoring functions to evaluate binding strengths. However, to meet the efficiency demands of SBVS, these algorithms and functions are often simplified, prioritizing speed over accuracy.</p><p >The emergence of deep learning (DL) has exerted a profound impact on diverse fields, ranging from natural language processing to computer vision and drug discovery. DeepMind’s AlphaFold2 has impressively exhibited its ability to accurately predict protein structures solely from amino acid sequences, highlighting the remarkable potential of DL in conformation prediction. This groundbreaking advancement circumvents the traditional search-scoring frameworks in LD, enhancing both accuracy and processing speed and thereby catalyzing a broader adoption of DL algorithms in binding pose prediction. Nevertheless, a consensus on certain aspects remains elusive.</p><p >In this Account, we delineate the current status of employing DL to augment LD within the VS paradigm, highlighting our contributions to this domain. Furthermore, we discuss the challenges and future prospects, drawing insights from our scholarly investigations. Initially, we present an overview of VS and LD, followed by an introduction to DL paradigms, which deviate significantly from traditional search-scoring frameworks. Subsequently, we delve into the challenges associated with the development of DL-based LD (DLLD), encompassing evaluation metrics, application scenarios, and physical plausibility of the predicted conformations. In the evaluation of LD algorithms, it is essential to recognize the multifaceted nature of the metrics. While the accuracy of binding pose prediction, often measured by the success rate, is a pivotal aspect, the scoring/screening power and computational speed of these algorithms are equally important given the pivotal role of LD tools in VS. Regarding application scenarios, early methods focused on blind docking, where the binding site is unknown. However, recent studies suggest a shift toward identifying binding sites rather than solely predicting binding poses within these models. In contrast, LD with a known pocket in VS has been shown to be more practical. Physical plausibility poses another significant challenge. Although DLLD models often achieve higher success rates compared to traditional methods, they may generate poses with implausible local structures, such as incorrect bond angles or lengths, which are disadvantageous for postprocessing tasks like visualization. Finally, we discuss the future perspectives for DLLD, emphasizing the need to improve generalization ability, strike a balance between speed and accuracy, account for protein conformation flexibility, and enhance physical plausibility. Additionally, we delve into the comparison between generative and regression algorithms in this context, exploring their respective strengths and potential.</p>\",\"PeriodicalId\":1,\"journal\":{\"name\":\"Accounts of Chemical Research\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":16.4000,\"publicationDate\":\"2024-04-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Accounts of Chemical Research\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.accounts.4c00093\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Accounts of Chemical Research","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.accounts.4c00093","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Advancing Ligand Docking through Deep Learning: Challenges and Prospects in Virtual Screening
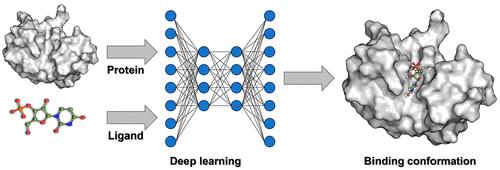
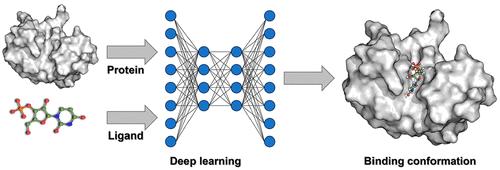
Molecular docking, also termed ligand docking (LD), is a pivotal element of structure-based virtual screening (SBVS) used to predict the binding conformations and affinities of protein–ligand complexes. Traditional LD methodologies rely on a search and scoring framework, utilizing heuristic algorithms to explore binding conformations and scoring functions to evaluate binding strengths. However, to meet the efficiency demands of SBVS, these algorithms and functions are often simplified, prioritizing speed over accuracy.
The emergence of deep learning (DL) has exerted a profound impact on diverse fields, ranging from natural language processing to computer vision and drug discovery. DeepMind’s AlphaFold2 has impressively exhibited its ability to accurately predict protein structures solely from amino acid sequences, highlighting the remarkable potential of DL in conformation prediction. This groundbreaking advancement circumvents the traditional search-scoring frameworks in LD, enhancing both accuracy and processing speed and thereby catalyzing a broader adoption of DL algorithms in binding pose prediction. Nevertheless, a consensus on certain aspects remains elusive.
In this Account, we delineate the current status of employing DL to augment LD within the VS paradigm, highlighting our contributions to this domain. Furthermore, we discuss the challenges and future prospects, drawing insights from our scholarly investigations. Initially, we present an overview of VS and LD, followed by an introduction to DL paradigms, which deviate significantly from traditional search-scoring frameworks. Subsequently, we delve into the challenges associated with the development of DL-based LD (DLLD), encompassing evaluation metrics, application scenarios, and physical plausibility of the predicted conformations. In the evaluation of LD algorithms, it is essential to recognize the multifaceted nature of the metrics. While the accuracy of binding pose prediction, often measured by the success rate, is a pivotal aspect, the scoring/screening power and computational speed of these algorithms are equally important given the pivotal role of LD tools in VS. Regarding application scenarios, early methods focused on blind docking, where the binding site is unknown. However, recent studies suggest a shift toward identifying binding sites rather than solely predicting binding poses within these models. In contrast, LD with a known pocket in VS has been shown to be more practical. Physical plausibility poses another significant challenge. Although DLLD models often achieve higher success rates compared to traditional methods, they may generate poses with implausible local structures, such as incorrect bond angles or lengths, which are disadvantageous for postprocessing tasks like visualization. Finally, we discuss the future perspectives for DLLD, emphasizing the need to improve generalization ability, strike a balance between speed and accuracy, account for protein conformation flexibility, and enhance physical plausibility. Additionally, we delve into the comparison between generative and regression algorithms in this context, exploring their respective strengths and potential.
期刊介绍:
Accounts of Chemical Research presents short, concise and critical articles offering easy-to-read overviews of basic research and applications in all areas of chemistry and biochemistry. These short reviews focus on research from the author’s own laboratory and are designed to teach the reader about a research project. In addition, Accounts of Chemical Research publishes commentaries that give an informed opinion on a current research problem. Special Issues online are devoted to a single topic of unusual activity and significance.
Accounts of Chemical Research replaces the traditional article abstract with an article "Conspectus." These entries synopsize the research affording the reader a closer look at the content and significance of an article. Through this provision of a more detailed description of the article contents, the Conspectus enhances the article's discoverability by search engines and the exposure for the research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
