{"title":"又有两个家族支持单基因脊髓小脑共济失调症的存在 48","authors":"Flavia Palombo, Alessandro Vaisfeld, Valentina Concetta Tropeano, Danara Ormanbekova, Isabelle Bacchi, Claudio Fiorini, Adelaide Peruzzi, Luca Morandi, Rocco Liguori, Valerio Carelli, Giovanni Rizzo","doi":"10.1007/s10048-024-00758-8","DOIUrl":null,"url":null,"abstract":"<p>The reduced penetrance of <i>TBP</i> intermediate alleles and the recently proposed possible digenic <i>TBP/STUB1</i> inheritance raised questions on the possible mechanism involved opening a debate on the existence of SCA48 as a monogenic disorder. We here report clinical and genetic results of two apparently unrelated patients carrying the same <i>STUB1</i> variant(c.244G > T;p.Asp82Tyr) with normal <i>TBP</i> alleles and a clinical picture fully resembling SCA48, including cerebellar ataxia, dysarthria and mild cognitive impairment. This report provides supportive evidence that this specific ataxia can also occur as a monogenic disease, considering classical <i>TBP</i> allelic ranges.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"3 1","pages":""},"PeriodicalIF":1.2000,"publicationDate":"2024-04-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Two more families supporting the existence of monogenic spinocerebellar ataxia 48\",\"authors\":\"Flavia Palombo, Alessandro Vaisfeld, Valentina Concetta Tropeano, Danara Ormanbekova, Isabelle Bacchi, Claudio Fiorini, Adelaide Peruzzi, Luca Morandi, Rocco Liguori, Valerio Carelli, Giovanni Rizzo\",\"doi\":\"10.1007/s10048-024-00758-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The reduced penetrance of <i>TBP</i> intermediate alleles and the recently proposed possible digenic <i>TBP/STUB1</i> inheritance raised questions on the possible mechanism involved opening a debate on the existence of SCA48 as a monogenic disorder. We here report clinical and genetic results of two apparently unrelated patients carrying the same <i>STUB1</i> variant(c.244G > T;p.Asp82Tyr) with normal <i>TBP</i> alleles and a clinical picture fully resembling SCA48, including cerebellar ataxia, dysarthria and mild cognitive impairment. This report provides supportive evidence that this specific ataxia can also occur as a monogenic disease, considering classical <i>TBP</i> allelic ranges.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\"3 1\",\"pages\":\"\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2024-04-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-024-00758-8\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-024-00758-8","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Two more families supporting the existence of monogenic spinocerebellar ataxia 48
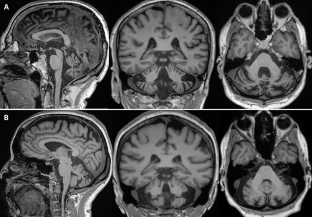
The reduced penetrance of TBP intermediate alleles and the recently proposed possible digenic TBP/STUB1 inheritance raised questions on the possible mechanism involved opening a debate on the existence of SCA48 as a monogenic disorder. We here report clinical and genetic results of two apparently unrelated patients carrying the same STUB1 variant(c.244G > T;p.Asp82Tyr) with normal TBP alleles and a clinical picture fully resembling SCA48, including cerebellar ataxia, dysarthria and mild cognitive impairment. This report provides supportive evidence that this specific ataxia can also occur as a monogenic disease, considering classical TBP allelic ranges.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
