L. Reginald Mills, Junho Kim, Eric M. Simmons, Steven R. Wisniewski and Paul J. Chirik*,
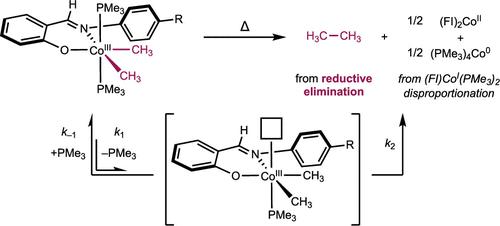
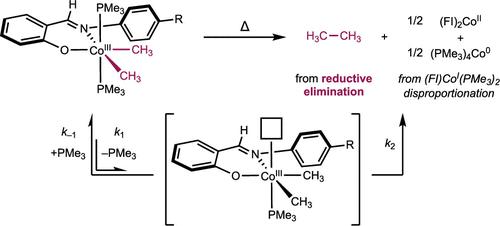
{"title":"苯氧亚氨基钴(III)(CH3)2(PMe3)2 配合物的 C(sp3)-C(sp3)还原消除作用","authors":"L. Reginald Mills, Junho Kim, Eric M. Simmons, Steven R. Wisniewski and Paul J. Chirik*, ","doi":"10.1021/acs.organomet.4c00070","DOIUrl":null,"url":null,"abstract":"<p >A series of six-coordinate, idealized octahedral phenoxyimine (FI)–cobalt(III) dimethyl bis(trimethylphosphine) complexes was synthesized and characterized by NMR spectroscopy and single-crystal X-ray diffraction. The thermal stability of the parent Ph-FI complex was evaluated at 60 °C in benzene-<i>d</i><sub>6</sub>, and a 13(1):1 ratio of ethane to methane was observed. The major detectable cobalt product of this reaction was the bis(chelate) cobalt derivative (Ph-FI)<sub>2</sub>Co formed by disproportionation of the (FI)cobalt(I) product following ethane reductive elimination. Addition of excess PMe<sub>3</sub> inhibited C(sp<sup>3</sup>)–C(sp<sup>3</sup>) reductive elimination, consistent with phosphine dissociation preceding C–C bond formation from a five-coordinate (FI)cobalt(III) dimethyl intermediate. The reductive elimination of substituted (R-FI)cobalt(III) dimethyl bis(trimethylphosphine) compounds was evaluated in acetonitrile-<i>d</i><sub>3</sub>, where ligands bearing electron-donating aniline substituents underwent reductive elimination the fastest and electron-withdrawing substituents the slowest. These data support a buildup of positive charge in the rate-limiting step, consistent with the formation of a more electropositive five-coordinate cobalt center prior to rate-limiting C–C reductive elimination. Attempted synthesis of a cobalt(III) dimethyl complex bearing a sterically hindered FI ligand with a <i>tert</i>-butyl substituent <i>ortho</i> to the phenol led exclusively to the corresponding bis(chelate) cobalt derivative, whose formation was rationalized by steric destabilization of pre–reductive elimination intermediates.</p>","PeriodicalId":56,"journal":{"name":"Organometallics","volume":"43 9","pages":"1021–1029"},"PeriodicalIF":2.9000,"publicationDate":"2024-04-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"C(sp3)–C(sp3) Reductive Elimination from (Phenoxyimine)Cobalt(III)(CH3)2(PMe3)2 Complexes\",\"authors\":\"L. Reginald Mills, Junho Kim, Eric M. Simmons, Steven R. Wisniewski and Paul J. Chirik*, \",\"doi\":\"10.1021/acs.organomet.4c00070\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >A series of six-coordinate, idealized octahedral phenoxyimine (FI)–cobalt(III) dimethyl bis(trimethylphosphine) complexes was synthesized and characterized by NMR spectroscopy and single-crystal X-ray diffraction. The thermal stability of the parent Ph-FI complex was evaluated at 60 °C in benzene-<i>d</i><sub>6</sub>, and a 13(1):1 ratio of ethane to methane was observed. The major detectable cobalt product of this reaction was the bis(chelate) cobalt derivative (Ph-FI)<sub>2</sub>Co formed by disproportionation of the (FI)cobalt(I) product following ethane reductive elimination. Addition of excess PMe<sub>3</sub> inhibited C(sp<sup>3</sup>)–C(sp<sup>3</sup>) reductive elimination, consistent with phosphine dissociation preceding C–C bond formation from a five-coordinate (FI)cobalt(III) dimethyl intermediate. The reductive elimination of substituted (R-FI)cobalt(III) dimethyl bis(trimethylphosphine) compounds was evaluated in acetonitrile-<i>d</i><sub>3</sub>, where ligands bearing electron-donating aniline substituents underwent reductive elimination the fastest and electron-withdrawing substituents the slowest. These data support a buildup of positive charge in the rate-limiting step, consistent with the formation of a more electropositive five-coordinate cobalt center prior to rate-limiting C–C reductive elimination. Attempted synthesis of a cobalt(III) dimethyl complex bearing a sterically hindered FI ligand with a <i>tert</i>-butyl substituent <i>ortho</i> to the phenol led exclusively to the corresponding bis(chelate) cobalt derivative, whose formation was rationalized by steric destabilization of pre–reductive elimination intermediates.</p>\",\"PeriodicalId\":56,\"journal\":{\"name\":\"Organometallics\",\"volume\":\"43 9\",\"pages\":\"1021–1029\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-04-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Organometallics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.organomet.4c00070\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Organometallics","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.organomet.4c00070","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
摘要
合成了一系列六配位理想化八面体苯氧亚胺(FI)-二甲基双(三甲基膦)钴(III)配合物,并通过核磁共振光谱和单晶 X 射线衍射对其进行了表征。在 60 °C 的苯-d6 中评估了母体 Ph-FI 复合物的热稳定性,观察到乙烷与甲烷的比例为 13(1):1。该反应中可检测到的主要钴产物是乙烷还原消除后,(FI)钴(I)产物歧化形成的双(螯合)钴衍生物 (Ph-FI)2Co。过量 PMe3 的加入抑制了 C(sp3)-C(sp3)的还原消除,这与五配位 (FI) 钴(III)二甲基中间体在形成 C-C 键之前膦解离是一致的。在乙腈-d3 中对取代的 (R-FI)钴(III)二甲基双(三甲基膦)化合物的还原消除进行了评估,在这些化合物中,带有捐电子苯胺取代基的配体的还原消除速度最快,而带有抽电子取代基的配体的还原消除速度最慢。这些数据支持在限速步骤中积累正电荷,这与在限速 C-C 还原消除之前形成电性更强的五配位钴中心相一致。在尝试合成一种钴(III)二甲基配合物时,发现该配合物带有一个立体受阻的 FI 配体,该配体在苯酚的正上方带有一个叔丁基取代基,因此只能得到相应的双(螯合物)钴衍生物,而这种钴衍生物的形成是由于还原消除前中间体的立体不稳定性造成的。
C(sp3)–C(sp3) Reductive Elimination from (Phenoxyimine)Cobalt(III)(CH3)2(PMe3)2 Complexes
A series of six-coordinate, idealized octahedral phenoxyimine (FI)–cobalt(III) dimethyl bis(trimethylphosphine) complexes was synthesized and characterized by NMR spectroscopy and single-crystal X-ray diffraction. The thermal stability of the parent Ph-FI complex was evaluated at 60 °C in benzene-d6, and a 13(1):1 ratio of ethane to methane was observed. The major detectable cobalt product of this reaction was the bis(chelate) cobalt derivative (Ph-FI)2Co formed by disproportionation of the (FI)cobalt(I) product following ethane reductive elimination. Addition of excess PMe3 inhibited C(sp3)–C(sp3) reductive elimination, consistent with phosphine dissociation preceding C–C bond formation from a five-coordinate (FI)cobalt(III) dimethyl intermediate. The reductive elimination of substituted (R-FI)cobalt(III) dimethyl bis(trimethylphosphine) compounds was evaluated in acetonitrile-d3, where ligands bearing electron-donating aniline substituents underwent reductive elimination the fastest and electron-withdrawing substituents the slowest. These data support a buildup of positive charge in the rate-limiting step, consistent with the formation of a more electropositive five-coordinate cobalt center prior to rate-limiting C–C reductive elimination. Attempted synthesis of a cobalt(III) dimethyl complex bearing a sterically hindered FI ligand with a tert-butyl substituent ortho to the phenol led exclusively to the corresponding bis(chelate) cobalt derivative, whose formation was rationalized by steric destabilization of pre–reductive elimination intermediates.
期刊介绍:
Organometallics is the flagship journal of organometallic chemistry and records progress in one of the most active fields of science, bridging organic and inorganic chemistry. The journal publishes Articles, Communications, Reviews, and Tutorials (instructional overviews) that depict research on the synthesis, structure, bonding, chemical reactivity, and reaction mechanisms for a variety of applications, including catalyst design and catalytic processes; main-group, transition-metal, and lanthanide and actinide metal chemistry; synthetic aspects of polymer science and materials science; and bioorganometallic chemistry.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
