Pierre-François Loos, Antoine Marie and Abdallah Ammar
{"title":"分子的累积格林函数方法","authors":"Pierre-François Loos, Antoine Marie and Abdallah Ammar","doi":"10.1039/D4FD00037D","DOIUrl":null,"url":null,"abstract":"<p >The cumulant expansion of the Green's function is a computationally efficient beyond-GW approach renowned for its significant enhancement of satellite features in materials. In contrast to the ubiquitous GW approximation of many-body perturbation theory, <em>ab initio</em> cumulant expansions performed on top of GW (GW + C) have demonstrated the capability to handle multi-particle processes by incorporating higher-order correlation effects or vertex corrections, yielding better agreements between experiment and theory for satellite structures. While widely employed in condensed matter physics, very few applications of GW + C have been published on molecular systems. Here, we assess the performance of this scheme on a series of 10-electron molecular systems (Ne, HF, H<small><sub>2</sub></small>O, NH<small><sub>3</sub></small>, and CH<small><sub>4</sub></small>) where full configuration interaction estimates of the outer-valence quasiparticle and satellite energies are available.</p>","PeriodicalId":49075,"journal":{"name":"Faraday Discussions","volume":"254 ","pages":" 240-260"},"PeriodicalIF":3.1000,"publicationDate":"2024-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Cumulant Green's function methods for molecules†\",\"authors\":\"Pierre-François Loos, Antoine Marie and Abdallah Ammar\",\"doi\":\"10.1039/D4FD00037D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The cumulant expansion of the Green's function is a computationally efficient beyond-GW approach renowned for its significant enhancement of satellite features in materials. In contrast to the ubiquitous GW approximation of many-body perturbation theory, <em>ab initio</em> cumulant expansions performed on top of GW (GW + C) have demonstrated the capability to handle multi-particle processes by incorporating higher-order correlation effects or vertex corrections, yielding better agreements between experiment and theory for satellite structures. While widely employed in condensed matter physics, very few applications of GW + C have been published on molecular systems. Here, we assess the performance of this scheme on a series of 10-electron molecular systems (Ne, HF, H<small><sub>2</sub></small>O, NH<small><sub>3</sub></small>, and CH<small><sub>4</sub></small>) where full configuration interaction estimates of the outer-valence quasiparticle and satellite energies are available.</p>\",\"PeriodicalId\":49075,\"journal\":{\"name\":\"Faraday Discussions\",\"volume\":\"254 \",\"pages\":\" 240-260\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-04-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Faraday Discussions\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00037d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Faraday Discussions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00037d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Chemistry","Score":null,"Total":0}
引用次数: 0
摘要
格林函数的累积展开是一种计算高效的超越全球定位系统的方法,因其显著增强了材料中的卫星特征而闻名。与多体扰动理论中无处不在的 GW 近似相比,在 GW(GW+C)基础上进行的 ab initio 积展开证明了通过纳入高阶相关效应或顶点校正来处理多粒子过程的能力,从而在卫星结构的实验与理论之间取得更好的一致。虽然 GW+C 广泛应用于凝聚态物理,但在分子系统中的应用却寥寥无几。在这里,我们评估了这一方案在一系列 10 电子分子系统(Ne、HF、H2O、NH3 和 CH4)上的性能,这些系统的外价准粒子和卫星能量的完全构型相互作用估计值是可用的。
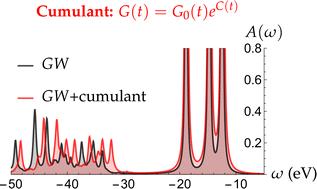
The cumulant expansion of the Green's function is a computationally efficient beyond-GW approach renowned for its significant enhancement of satellite features in materials. In contrast to the ubiquitous GW approximation of many-body perturbation theory, ab initio cumulant expansions performed on top of GW (GW + C) have demonstrated the capability to handle multi-particle processes by incorporating higher-order correlation effects or vertex corrections, yielding better agreements between experiment and theory for satellite structures. While widely employed in condensed matter physics, very few applications of GW + C have been published on molecular systems. Here, we assess the performance of this scheme on a series of 10-electron molecular systems (Ne, HF, H2O, NH3, and CH4) where full configuration interaction estimates of the outer-valence quasiparticle and satellite energies are available.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
