Melissa Pauly, Mandy Krumbiegel, Sandra Trumpp, Sonja Braig, Thomas Rupprecht, Cornelia Kraus, Steffen Uebe, André Reis, Georgia Vasileiou
{"title":"与 CTNND2 双重复缺失相关的劳赫-阿扎雷洛综合征的严重表现","authors":"Melissa Pauly, Mandy Krumbiegel, Sandra Trumpp, Sonja Braig, Thomas Rupprecht, Cornelia Kraus, Steffen Uebe, André Reis, Georgia Vasileiou","doi":"10.1111/cge.14532","DOIUrl":null,"url":null,"abstract":"<p><i>CTNND2</i> encodes δ-catenin, a component of an adherens junction complex, and plays an important role in neuronal structure and function. To date, only heterozygous loss-of-function <i>CTNND2</i> variants have been associated with mild neurodevelopmental delay and behavioral anomalies, a condition, which we named Rauch-Azzarello syndrome. Here, we report three siblings of a consanguineous family of Syrian descent with a homozygous deletion encompassing the last 19 exons of <i>CTNND2</i> predicted to disrupt the transcript. All presented with severe neurodevelopmental delay with absent speech, profound motor delay, stereotypic behavior, microcephaly, short stature, muscular hypotonia with lower limb hypertonia, and variable eye anomalies. The parents and the fourth sibling were heterozygous carriers of the deletion and exhibited mild neurodevelopmental impairment resembling that of the previously described heterozygous individuals. The present study unveils a severe manifestation of <i>CTNND2</i>-associated Rauch-Azzarello syndrome attributed to biallelic loss-of-function aberrations, clinically distinct from the already described mild presentation of heterozygous individuals. Furthermore, we demonstrate novel clinical features in homozygous individuals that have not been reported in heterozygous cases to date.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":null,"pages":null},"PeriodicalIF":2.9000,"publicationDate":"2024-04-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14532","citationCount":"0","resultStr":"{\"title\":\"Severe manifestation of Rauch-Azzarello syndrome associated with biallelic deletion of CTNND2\",\"authors\":\"Melissa Pauly, Mandy Krumbiegel, Sandra Trumpp, Sonja Braig, Thomas Rupprecht, Cornelia Kraus, Steffen Uebe, André Reis, Georgia Vasileiou\",\"doi\":\"10.1111/cge.14532\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><i>CTNND2</i> encodes δ-catenin, a component of an adherens junction complex, and plays an important role in neuronal structure and function. To date, only heterozygous loss-of-function <i>CTNND2</i> variants have been associated with mild neurodevelopmental delay and behavioral anomalies, a condition, which we named Rauch-Azzarello syndrome. Here, we report three siblings of a consanguineous family of Syrian descent with a homozygous deletion encompassing the last 19 exons of <i>CTNND2</i> predicted to disrupt the transcript. All presented with severe neurodevelopmental delay with absent speech, profound motor delay, stereotypic behavior, microcephaly, short stature, muscular hypotonia with lower limb hypertonia, and variable eye anomalies. The parents and the fourth sibling were heterozygous carriers of the deletion and exhibited mild neurodevelopmental impairment resembling that of the previously described heterozygous individuals. The present study unveils a severe manifestation of <i>CTNND2</i>-associated Rauch-Azzarello syndrome attributed to biallelic loss-of-function aberrations, clinically distinct from the already described mild presentation of heterozygous individuals. Furthermore, we demonstrate novel clinical features in homozygous individuals that have not been reported in heterozygous cases to date.</p>\",\"PeriodicalId\":10354,\"journal\":{\"name\":\"Clinical Genetics\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-04-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14532\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cge.14532\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14532","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Severe manifestation of Rauch-Azzarello syndrome associated with biallelic deletion of CTNND2
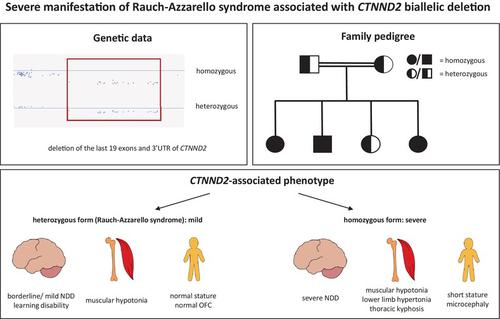
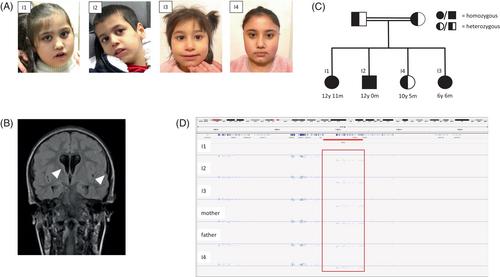
CTNND2 encodes δ-catenin, a component of an adherens junction complex, and plays an important role in neuronal structure and function. To date, only heterozygous loss-of-function CTNND2 variants have been associated with mild neurodevelopmental delay and behavioral anomalies, a condition, which we named Rauch-Azzarello syndrome. Here, we report three siblings of a consanguineous family of Syrian descent with a homozygous deletion encompassing the last 19 exons of CTNND2 predicted to disrupt the transcript. All presented with severe neurodevelopmental delay with absent speech, profound motor delay, stereotypic behavior, microcephaly, short stature, muscular hypotonia with lower limb hypertonia, and variable eye anomalies. The parents and the fourth sibling were heterozygous carriers of the deletion and exhibited mild neurodevelopmental impairment resembling that of the previously described heterozygous individuals. The present study unveils a severe manifestation of CTNND2-associated Rauch-Azzarello syndrome attributed to biallelic loss-of-function aberrations, clinically distinct from the already described mild presentation of heterozygous individuals. Furthermore, we demonstrate novel clinical features in homozygous individuals that have not been reported in heterozygous cases to date.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
