Qianjin Liu, Lijin Jiao, Mao-Sen Ye, Zhiyu Ma, Jinsong Yu, Ling-Yan Su, Wei-Yin Zou, Lu-Xiu Yang, Chang Chen, Yong-Gang Yao
{"title":"GSNOR 通过 S-亚硝基化 MAPK14 负向调节 NLRP3 炎症体","authors":"Qianjin Liu, Lijin Jiao, Mao-Sen Ye, Zhiyu Ma, Jinsong Yu, Ling-Yan Su, Wei-Yin Zou, Lu-Xiu Yang, Chang Chen, Yong-Gang Yao","doi":"10.1038/s41423-024-01155-9","DOIUrl":null,"url":null,"abstract":"Hyperactivation of the NLRP3 inflammasome has been implicated in the pathogenesis of numerous diseases. However, the precise molecular mechanisms that modulate the transcriptional regulation of NLRP3 remain largely unknown. In this study, we demonstrated that S-nitrosoglutathione reductase (GSNOR) deficiency in macrophages leads to significant increases in the Nlrp3 and Il-1β expression levels and interleukin-1β (IL-1β) secretion in response to NLRP3 inflammasome stimulation. Furthermore, in vivo experiments utilizing Gsnor−/− mice revealed increased disease severity in both lipopolysaccharide (LPS)-induced septic shock and dextran sodium sulfate (DSS)-induced colitis models. Additionally, we showed that both LPS-induced septic shock and DSS-induced colitis were ameliorated in Gsnor−/− Nlrp3−/− double-knockout (DKO) mice. Mechanistically, GSNOR deficiency increases the S-nitrosation of mitogen-activated protein kinase 14 (MAPK14) at the Cys211 residue and augments MAPK14 kinase activity, thereby promoting Nlrp3 and Il-1β transcription and stimulating NLRP3 inflammasome activity. Our findings suggested that GSNOR is a regulator of the NLRP3 inflammasome and that reducing the level of S-nitrosylated MAPK14 may constitute an effective strategy for alleviating diseases associated with NLRP3-mediated inflammation.","PeriodicalId":9950,"journal":{"name":"Cellular &Molecular Immunology","volume":"21 6","pages":"561-574"},"PeriodicalIF":23.9000,"publicationDate":"2024-04-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"GSNOR negatively regulates the NLRP3 inflammasome via S-nitrosation of MAPK14\",\"authors\":\"Qianjin Liu, Lijin Jiao, Mao-Sen Ye, Zhiyu Ma, Jinsong Yu, Ling-Yan Su, Wei-Yin Zou, Lu-Xiu Yang, Chang Chen, Yong-Gang Yao\",\"doi\":\"10.1038/s41423-024-01155-9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Hyperactivation of the NLRP3 inflammasome has been implicated in the pathogenesis of numerous diseases. However, the precise molecular mechanisms that modulate the transcriptional regulation of NLRP3 remain largely unknown. In this study, we demonstrated that S-nitrosoglutathione reductase (GSNOR) deficiency in macrophages leads to significant increases in the Nlrp3 and Il-1β expression levels and interleukin-1β (IL-1β) secretion in response to NLRP3 inflammasome stimulation. Furthermore, in vivo experiments utilizing Gsnor−/− mice revealed increased disease severity in both lipopolysaccharide (LPS)-induced septic shock and dextran sodium sulfate (DSS)-induced colitis models. Additionally, we showed that both LPS-induced septic shock and DSS-induced colitis were ameliorated in Gsnor−/− Nlrp3−/− double-knockout (DKO) mice. Mechanistically, GSNOR deficiency increases the S-nitrosation of mitogen-activated protein kinase 14 (MAPK14) at the Cys211 residue and augments MAPK14 kinase activity, thereby promoting Nlrp3 and Il-1β transcription and stimulating NLRP3 inflammasome activity. Our findings suggested that GSNOR is a regulator of the NLRP3 inflammasome and that reducing the level of S-nitrosylated MAPK14 may constitute an effective strategy for alleviating diseases associated with NLRP3-mediated inflammation.\",\"PeriodicalId\":9950,\"journal\":{\"name\":\"Cellular &Molecular Immunology\",\"volume\":\"21 6\",\"pages\":\"561-574\"},\"PeriodicalIF\":23.9000,\"publicationDate\":\"2024-04-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cellular &Molecular Immunology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.nature.com/articles/s41423-024-01155-9\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cellular &Molecular Immunology","FirstCategoryId":"3","ListUrlMain":"https://www.nature.com/articles/s41423-024-01155-9","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
GSNOR negatively regulates the NLRP3 inflammasome via S-nitrosation of MAPK14
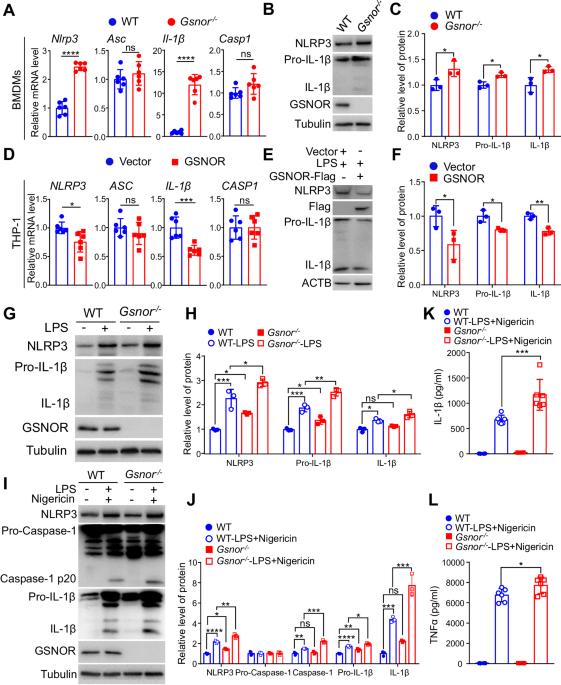
Hyperactivation of the NLRP3 inflammasome has been implicated in the pathogenesis of numerous diseases. However, the precise molecular mechanisms that modulate the transcriptional regulation of NLRP3 remain largely unknown. In this study, we demonstrated that S-nitrosoglutathione reductase (GSNOR) deficiency in macrophages leads to significant increases in the Nlrp3 and Il-1β expression levels and interleukin-1β (IL-1β) secretion in response to NLRP3 inflammasome stimulation. Furthermore, in vivo experiments utilizing Gsnor−/− mice revealed increased disease severity in both lipopolysaccharide (LPS)-induced septic shock and dextran sodium sulfate (DSS)-induced colitis models. Additionally, we showed that both LPS-induced septic shock and DSS-induced colitis were ameliorated in Gsnor−/− Nlrp3−/− double-knockout (DKO) mice. Mechanistically, GSNOR deficiency increases the S-nitrosation of mitogen-activated protein kinase 14 (MAPK14) at the Cys211 residue and augments MAPK14 kinase activity, thereby promoting Nlrp3 and Il-1β transcription and stimulating NLRP3 inflammasome activity. Our findings suggested that GSNOR is a regulator of the NLRP3 inflammasome and that reducing the level of S-nitrosylated MAPK14 may constitute an effective strategy for alleviating diseases associated with NLRP3-mediated inflammation.
期刊介绍:
Cellular & Molecular Immunology, a monthly journal from the Chinese Society of Immunology and the University of Science and Technology of China, serves as a comprehensive platform covering both basic immunology research and clinical applications. The journal publishes a variety of article types, including Articles, Review Articles, Mini Reviews, and Short Communications, focusing on diverse aspects of cellular and molecular immunology.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
