Felix Schmalz, Wassja A. Kopp, Eirini Goudeli, Kai Leonhard
{"title":"从碳氢化合物热解的分子动力学模拟中识别和验证反应路径","authors":"Felix Schmalz, Wassja A. Kopp, Eirini Goudeli, Kai Leonhard","doi":"10.1002/kin.21719","DOIUrl":null,"url":null,"abstract":"<p>Creation of complex chemical mechanisms for hydrocarbon pyrolysis and combustion is challenging due to the large number of species and reactions involved. Reactive molecular dynamics (RMD) enables the simulation of thousands of reactions and the discovery of previously unknown components of the reaction network. However, due to the inherent imprecision of reactive force fields, it is necessary to verify RMD-obtained reaction paths using more accurate methods such as Density Functional Theory (DFT). We demonstrate a method for identification and confirmation of reaction pathways from RMD that supplement an established mechanism, using the example of benzene formation from <i>n</i>-heptane and <i>iso</i>-octane pyrolysis. We establish a validation workflow to extract reaction geometries from RMD and optimize transition states using the Nudged-Elastic-Band method on semi-empirical and quantum mechanical levels of theory. Our findings demonstrate that the widely recognized ReaxFF parameterization, CHO2016, can identify known pathways from a established soot formation mechanism while also indicating new ones. We also show that CHO2016 underestimates hydrogen migration barriers by up to <span></span><math>\n <semantics>\n <mrow>\n <mn>40</mn>\n <mspace></mspace>\n <msup>\n <mrow>\n <mi>kcal</mi>\n <mspace></mspace>\n <mi>mol</mi>\n </mrow>\n <mrow>\n <mo>−</mo>\n <mn>1</mn>\n </mrow>\n </msup>\n </mrow>\n <annotation>$40\\,{\\rm {kcal\\,mol}}^{-1}$</annotation>\n </semantics></math> as compared to DFT and can lower activation barriers significantly for spin-forbidden reactions. This highlights the necessity for validation or potentially even reparametrization of CHO2016.</p>","PeriodicalId":13894,"journal":{"name":"International Journal of Chemical Kinetics","volume":"56 9","pages":"501-512"},"PeriodicalIF":1.6000,"publicationDate":"2024-04-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/kin.21719","citationCount":"0","resultStr":"{\"title\":\"Reaction path identification and validation from molecular dynamics simulations of hydrocarbon pyrolysis\",\"authors\":\"Felix Schmalz, Wassja A. Kopp, Eirini Goudeli, Kai Leonhard\",\"doi\":\"10.1002/kin.21719\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Creation of complex chemical mechanisms for hydrocarbon pyrolysis and combustion is challenging due to the large number of species and reactions involved. Reactive molecular dynamics (RMD) enables the simulation of thousands of reactions and the discovery of previously unknown components of the reaction network. However, due to the inherent imprecision of reactive force fields, it is necessary to verify RMD-obtained reaction paths using more accurate methods such as Density Functional Theory (DFT). We demonstrate a method for identification and confirmation of reaction pathways from RMD that supplement an established mechanism, using the example of benzene formation from <i>n</i>-heptane and <i>iso</i>-octane pyrolysis. We establish a validation workflow to extract reaction geometries from RMD and optimize transition states using the Nudged-Elastic-Band method on semi-empirical and quantum mechanical levels of theory. Our findings demonstrate that the widely recognized ReaxFF parameterization, CHO2016, can identify known pathways from a established soot formation mechanism while also indicating new ones. We also show that CHO2016 underestimates hydrogen migration barriers by up to <span></span><math>\\n <semantics>\\n <mrow>\\n <mn>40</mn>\\n <mspace></mspace>\\n <msup>\\n <mrow>\\n <mi>kcal</mi>\\n <mspace></mspace>\\n <mi>mol</mi>\\n </mrow>\\n <mrow>\\n <mo>−</mo>\\n <mn>1</mn>\\n </mrow>\\n </msup>\\n </mrow>\\n <annotation>$40\\\\,{\\\\rm {kcal\\\\,mol}}^{-1}$</annotation>\\n </semantics></math> as compared to DFT and can lower activation barriers significantly for spin-forbidden reactions. This highlights the necessity for validation or potentially even reparametrization of CHO2016.</p>\",\"PeriodicalId\":13894,\"journal\":{\"name\":\"International Journal of Chemical Kinetics\",\"volume\":\"56 9\",\"pages\":\"501-512\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2024-04-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/kin.21719\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Chemical Kinetics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/kin.21719\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Chemical Kinetics","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/kin.21719","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Reaction path identification and validation from molecular dynamics simulations of hydrocarbon pyrolysis
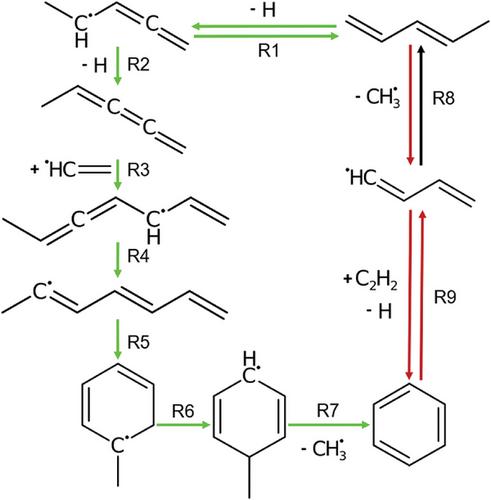
Creation of complex chemical mechanisms for hydrocarbon pyrolysis and combustion is challenging due to the large number of species and reactions involved. Reactive molecular dynamics (RMD) enables the simulation of thousands of reactions and the discovery of previously unknown components of the reaction network. However, due to the inherent imprecision of reactive force fields, it is necessary to verify RMD-obtained reaction paths using more accurate methods such as Density Functional Theory (DFT). We demonstrate a method for identification and confirmation of reaction pathways from RMD that supplement an established mechanism, using the example of benzene formation from n-heptane and iso-octane pyrolysis. We establish a validation workflow to extract reaction geometries from RMD and optimize transition states using the Nudged-Elastic-Band method on semi-empirical and quantum mechanical levels of theory. Our findings demonstrate that the widely recognized ReaxFF parameterization, CHO2016, can identify known pathways from a established soot formation mechanism while also indicating new ones. We also show that CHO2016 underestimates hydrogen migration barriers by up to as compared to DFT and can lower activation barriers significantly for spin-forbidden reactions. This highlights the necessity for validation or potentially even reparametrization of CHO2016.
期刊介绍:
As the leading archival journal devoted exclusively to chemical kinetics, the International Journal of Chemical Kinetics publishes original research in gas phase, condensed phase, and polymer reaction kinetics, as well as biochemical and surface kinetics. The Journal seeks to be the primary archive for careful experimental measurements of reaction kinetics, in both simple and complex systems. The Journal also presents new developments in applied theoretical kinetics and publishes large kinetic models, and the algorithms and estimates used in these models. These include methods for handling the large reaction networks important in biochemistry, catalysis, and free radical chemistry. In addition, the Journal explores such topics as the quantitative relationships between molecular structure and chemical reactivity, organic/inorganic chemistry and reaction mechanisms, and the reactive chemistry at interfaces.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
