{"title":"原发性甲状旁腺功能亢进症的家族性:最新进展","authors":"F. Cetani, E. Dinoi, L. Pierotti, E. Pardi","doi":"10.1007/s40618-024-02366-7","DOIUrl":null,"url":null,"abstract":"<h3 data-test=\"abstract-sub-heading\">Background</h3><p>Familial primary hyperparathyroidism (PHPT) includes syndromic and non-syndromic disorders. The former are characterized by the occurrence of PHPT in association with extra-parathyroid manifestations and includes multiple endocrine neoplasia (MEN) types 1, 2, and 4 syndromes, and hyperparathyroidism–jaw tumor (HPT–JT). The latter consists of familial hypocalciuric hypercalcemia (FHH) types 1, 2 and 3, neonatal severe primary hyperparathyroidism (NSHPT), and familial isolated primary hyperparathyroidism (FIHP). The familial forms of PHPT show different levels of PHPT penetrance, developing earlier and with multiglandular involvement compared to sporadic counterpart.</p><p>All these diseases exhibit Mendelian inheritance patterns, and for most of them, the genes responsible have been identified. DNA testing for predisposing mutations is helpful in index cases or in individuals with a high suspicion of the disease. Early recognition of hereditary disorders of PHPT is of great importance for the best clinical and surgical approach. Genetic testing is useful in routine clinical practice because it will also involve appropriate screening for extra-parathyroidal manifestations related to the syndrome as well as the identification of asymptomatic carriers of the mutation.</p><h3 data-test=\"abstract-sub-heading\">Purpose</h3><p>The aim of the review is to discuss the current knowledge on the clinical and genetic profile of these disorders along with the importance of genetic testing in clinical practice.</p>","PeriodicalId":15651,"journal":{"name":"Journal of Endocrinological Investigation","volume":"26 1","pages":""},"PeriodicalIF":3.5000,"publicationDate":"2024-04-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Familial states of primary hyperparathyroidism: an update\",\"authors\":\"F. Cetani, E. Dinoi, L. Pierotti, E. Pardi\",\"doi\":\"10.1007/s40618-024-02366-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<h3 data-test=\\\"abstract-sub-heading\\\">Background</h3><p>Familial primary hyperparathyroidism (PHPT) includes syndromic and non-syndromic disorders. The former are characterized by the occurrence of PHPT in association with extra-parathyroid manifestations and includes multiple endocrine neoplasia (MEN) types 1, 2, and 4 syndromes, and hyperparathyroidism–jaw tumor (HPT–JT). The latter consists of familial hypocalciuric hypercalcemia (FHH) types 1, 2 and 3, neonatal severe primary hyperparathyroidism (NSHPT), and familial isolated primary hyperparathyroidism (FIHP). The familial forms of PHPT show different levels of PHPT penetrance, developing earlier and with multiglandular involvement compared to sporadic counterpart.</p><p>All these diseases exhibit Mendelian inheritance patterns, and for most of them, the genes responsible have been identified. DNA testing for predisposing mutations is helpful in index cases or in individuals with a high suspicion of the disease. Early recognition of hereditary disorders of PHPT is of great importance for the best clinical and surgical approach. Genetic testing is useful in routine clinical practice because it will also involve appropriate screening for extra-parathyroidal manifestations related to the syndrome as well as the identification of asymptomatic carriers of the mutation.</p><h3 data-test=\\\"abstract-sub-heading\\\">Purpose</h3><p>The aim of the review is to discuss the current knowledge on the clinical and genetic profile of these disorders along with the importance of genetic testing in clinical practice.</p>\",\"PeriodicalId\":15651,\"journal\":{\"name\":\"Journal of Endocrinological Investigation\",\"volume\":\"26 1\",\"pages\":\"\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-04-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Endocrinological Investigation\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s40618-024-02366-7\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Endocrinological Investigation","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s40618-024-02366-7","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
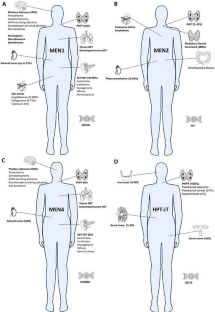
背景家族性原发性甲状旁腺功能亢进症(PHPT)包括综合征和非综合征疾病。前者的特点是PHPT的发生与甲状旁腺外表现有关,包括多发性内分泌肿瘤(MEN)1、2和4型综合征以及甲状旁腺功能亢进-下颌骨肿瘤(HPT-JT)。后者包括家族性低钙尿症高钙血症(FHH)1、2和3型、新生儿严重原发性甲状旁腺功能亢进症(NSHPT)和家族性孤立原发性甲状旁腺功能亢进症(FIHP)。与散发性原发性甲状旁腺功能亢进症相比,家族性原发性甲状旁腺功能亢进症表现出不同程度的PHPT渗透性,发病较早且多腺受累。对指数病例或高度怀疑患病的人进行 DNA 检测,有助于发现易感基因突变。及早发现 PHPT 的遗传性疾病对于采取最佳的临床和手术治疗方法非常重要。基因检测在常规临床实践中非常有用,因为它还能对与该综合征相关的甲状旁腺外表现进行适当筛查,并识别无症状的基因突变携带者。
Familial states of primary hyperparathyroidism: an update
Background
Familial primary hyperparathyroidism (PHPT) includes syndromic and non-syndromic disorders. The former are characterized by the occurrence of PHPT in association with extra-parathyroid manifestations and includes multiple endocrine neoplasia (MEN) types 1, 2, and 4 syndromes, and hyperparathyroidism–jaw tumor (HPT–JT). The latter consists of familial hypocalciuric hypercalcemia (FHH) types 1, 2 and 3, neonatal severe primary hyperparathyroidism (NSHPT), and familial isolated primary hyperparathyroidism (FIHP). The familial forms of PHPT show different levels of PHPT penetrance, developing earlier and with multiglandular involvement compared to sporadic counterpart.
All these diseases exhibit Mendelian inheritance patterns, and for most of them, the genes responsible have been identified. DNA testing for predisposing mutations is helpful in index cases or in individuals with a high suspicion of the disease. Early recognition of hereditary disorders of PHPT is of great importance for the best clinical and surgical approach. Genetic testing is useful in routine clinical practice because it will also involve appropriate screening for extra-parathyroidal manifestations related to the syndrome as well as the identification of asymptomatic carriers of the mutation.
Purpose
The aim of the review is to discuss the current knowledge on the clinical and genetic profile of these disorders along with the importance of genetic testing in clinical practice.
期刊介绍:
The Journal of Endocrinological Investigation is a well-established, e-only endocrine journal founded 36 years ago in 1978. It is the official journal of the Italian Society of Endocrinology (SIE), established in 1964. Other Italian societies in the endocrinology and metabolism field are affiliated to the journal: Italian Society of Andrology and Sexual Medicine, Italian Society of Obesity, Italian Society of Pediatric Endocrinology and Diabetology, Clinical Endocrinologists’ Association, Thyroid Association, Endocrine Surgical Units Association, Italian Society of Pharmacology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
