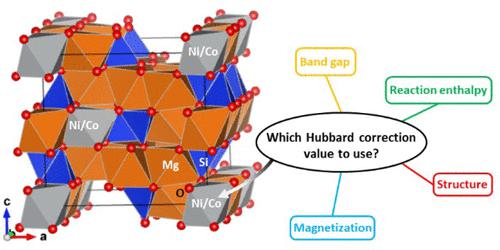
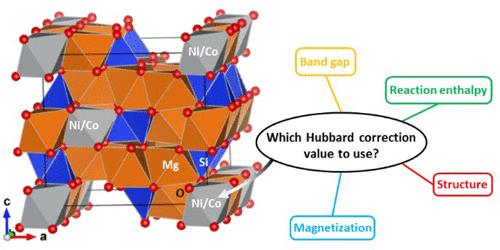
{"title":"福斯特石中的镍和钴掺杂:采用哈伯德校正的密度泛函理论研究","authors":"Michel Sassi, and , Sebastien N. Kerisit*, ","doi":"10.1021/acsearthspacechem.3c00370","DOIUrl":null,"url":null,"abstract":"<p >Ni and Co are critical elements for the world economy and modern technologies. Mafic and ultramafic deposits represent low-grade yet abundant alternatives to traditional Ni and Co ores. In this work, density functional theory (DFT) with the Hubbard <i>U</i> correction (DFT+<i>U</i>) was used to simulate the incorporation of Ni and Co in forsterite (Mg<sub>2</sub>SiO<sub>4</sub>), the Mg endmember of olivine, a common mineral in mafic and ultramafic rocks. Hubbard <i>U</i> terms for Ni and Co were parametrized using a series of oxide, hydroxide, carbonate, silicate, and sulfide minerals relevant to extraction and recovery of Ni and Co from mafic and ultramafic deposits. Electronic, energetic, magnetic, and structural properties were considered in the parametrization. For each of Ni and Co, an effective Hubbard correction (<i>U</i><sub>eff</sub>) value that optimized agreement with either experimental data or a hybrid exchange-correlation functional for all of the minerals considered is reported. DFT+<i>U</i> ab initio molecular dynamics (AIMD) simulations of Ni and Co incorporated into the M1 and M2 octahedral sites of forsterite were then performed. Ni and Co substitution in the M1 site was more energetically favorable than substitution in the M2 site, in agreement with published partition coefficients. AIMD trajectories were used to compute extended X-ray absorption fine structure (EXAFS) spectra of Ni in the M1 and M2 sites for direct fitting to a published experimental spectrum of Ni in a natural San Carlos olivine sample. The results of the fit indicated that ordering of Ni in the M1 site was not as strong at the low Ni concentrations relevant to mafic and ultramafic silicate minerals as that at the higher concentrations of the Ni-Mg olivine solid solutions studied to date.</p>","PeriodicalId":15,"journal":{"name":"ACS Earth and Space Chemistry","volume":"8 5","pages":"1027–1038"},"PeriodicalIF":2.9000,"publicationDate":"2024-04-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Ni and Co Incorporation in Forsterite: A Density Functional Theory Study with Hubbard Correction\",\"authors\":\"Michel Sassi, and , Sebastien N. Kerisit*, \",\"doi\":\"10.1021/acsearthspacechem.3c00370\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Ni and Co are critical elements for the world economy and modern technologies. Mafic and ultramafic deposits represent low-grade yet abundant alternatives to traditional Ni and Co ores. In this work, density functional theory (DFT) with the Hubbard <i>U</i> correction (DFT+<i>U</i>) was used to simulate the incorporation of Ni and Co in forsterite (Mg<sub>2</sub>SiO<sub>4</sub>), the Mg endmember of olivine, a common mineral in mafic and ultramafic rocks. Hubbard <i>U</i> terms for Ni and Co were parametrized using a series of oxide, hydroxide, carbonate, silicate, and sulfide minerals relevant to extraction and recovery of Ni and Co from mafic and ultramafic deposits. Electronic, energetic, magnetic, and structural properties were considered in the parametrization. For each of Ni and Co, an effective Hubbard correction (<i>U</i><sub>eff</sub>) value that optimized agreement with either experimental data or a hybrid exchange-correlation functional for all of the minerals considered is reported. DFT+<i>U</i> ab initio molecular dynamics (AIMD) simulations of Ni and Co incorporated into the M1 and M2 octahedral sites of forsterite were then performed. Ni and Co substitution in the M1 site was more energetically favorable than substitution in the M2 site, in agreement with published partition coefficients. AIMD trajectories were used to compute extended X-ray absorption fine structure (EXAFS) spectra of Ni in the M1 and M2 sites for direct fitting to a published experimental spectrum of Ni in a natural San Carlos olivine sample. The results of the fit indicated that ordering of Ni in the M1 site was not as strong at the low Ni concentrations relevant to mafic and ultramafic silicate minerals as that at the higher concentrations of the Ni-Mg olivine solid solutions studied to date.</p>\",\"PeriodicalId\":15,\"journal\":{\"name\":\"ACS Earth and Space Chemistry\",\"volume\":\"8 5\",\"pages\":\"1027–1038\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-04-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Earth and Space Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acsearthspacechem.3c00370\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Earth and Space Chemistry","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsearthspacechem.3c00370","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
镍和钴是世界经济和现代技术的关键元素。黑云母和超黑云母矿床是传统镍和钴矿石的低品位但丰富的替代品。本研究采用密度泛函理论(DFT)和哈伯德 U 修正(DFT+U)来模拟镍和钴在橄榄石(Mg2SiO4)中的掺入,橄榄石是岩浆岩和超岩浆岩中常见的矿物。利用一系列与从黑云母和超黑云母矿床中提取和回收镍和钴有关的氧化物、氢氧化物、碳酸盐、硅酸盐和硫化物矿物,对镍和钴的哈伯德 U 项进行了参数化。在参数化过程中考虑了电子、能量、磁性和结构特性。对于每种镍和钴,都报告了一个有效哈伯德校正(Ueff)值,该值优化了与实验数据或混合交换相关函数的一致性,适用于所考虑的所有矿物。然后,对镍和钴掺入绿柱石 M1 和 M2 八面体位点的 DFT+U 原子分子动力学(AIMD)模拟进行了分析。镍和钴在 M1 位点的取代比在 M2 位点的取代在能量上更有利,这与已公布的分配系数一致。利用 AIMD 轨迹计算了 M1 和 M2 位点中 Ni 的扩展 X 射线吸收精细结构 (EXAFS) 光谱,以便直接拟合已发表的天然圣卡洛斯橄榄石样本中 Ni 的实验光谱。拟合结果表明,在与黑云母和超黑云母硅酸盐矿物相关的低镍浓度下,M1 位点的镍有序性不如迄今为止研究的高浓度镍镁橄榄石固溶体中的镍有序性强。
Ni and Co Incorporation in Forsterite: A Density Functional Theory Study with Hubbard Correction
Ni and Co are critical elements for the world economy and modern technologies. Mafic and ultramafic deposits represent low-grade yet abundant alternatives to traditional Ni and Co ores. In this work, density functional theory (DFT) with the Hubbard U correction (DFT+U) was used to simulate the incorporation of Ni and Co in forsterite (Mg2SiO4), the Mg endmember of olivine, a common mineral in mafic and ultramafic rocks. Hubbard U terms for Ni and Co were parametrized using a series of oxide, hydroxide, carbonate, silicate, and sulfide minerals relevant to extraction and recovery of Ni and Co from mafic and ultramafic deposits. Electronic, energetic, magnetic, and structural properties were considered in the parametrization. For each of Ni and Co, an effective Hubbard correction (Ueff) value that optimized agreement with either experimental data or a hybrid exchange-correlation functional for all of the minerals considered is reported. DFT+U ab initio molecular dynamics (AIMD) simulations of Ni and Co incorporated into the M1 and M2 octahedral sites of forsterite were then performed. Ni and Co substitution in the M1 site was more energetically favorable than substitution in the M2 site, in agreement with published partition coefficients. AIMD trajectories were used to compute extended X-ray absorption fine structure (EXAFS) spectra of Ni in the M1 and M2 sites for direct fitting to a published experimental spectrum of Ni in a natural San Carlos olivine sample. The results of the fit indicated that ordering of Ni in the M1 site was not as strong at the low Ni concentrations relevant to mafic and ultramafic silicate minerals as that at the higher concentrations of the Ni-Mg olivine solid solutions studied to date.
期刊介绍:
The scope of ACS Earth and Space Chemistry includes the application of analytical, experimental and theoretical chemistry to investigate research questions relevant to the Earth and Space. The journal encompasses the highly interdisciplinary nature of research in this area, while emphasizing chemistry and chemical research tools as the unifying theme. The journal publishes broadly in the domains of high- and low-temperature geochemistry, atmospheric chemistry, marine chemistry, planetary chemistry, astrochemistry, and analytical geochemistry. ACS Earth and Space Chemistry publishes Articles, Letters, Reviews, and Features to provide flexible formats to readily communicate all aspects of research in these fields.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
