Razieh Khoshnevisan, Shakiba Hassanzadeh, Christoph Klein, Meino Rohlfs, Bodo Grimbacher, Newsha Molavi, Aryana Zamanifar, Ali Khoshnevisan, Mahbube Jafari, Bahram Bagherpour, Mahdiyeh Behnam, Somayeh Najafi, Roya Sherkat
{"title":"被诊断为先天性免疫错误患者的 B 细胞缺失:一项基于登记的研究","authors":"Razieh Khoshnevisan, Shakiba Hassanzadeh, Christoph Klein, Meino Rohlfs, Bodo Grimbacher, Newsha Molavi, Aryana Zamanifar, Ali Khoshnevisan, Mahbube Jafari, Bahram Bagherpour, Mahdiyeh Behnam, Somayeh Najafi, Roya Sherkat","doi":"10.1007/s00251-024-01342-y","DOIUrl":null,"url":null,"abstract":"<p>Hypogammaglobulinemia without B-cells is a subgroup of inborn errors of immunity (IEI) which is characterized by a significant decline in all serum immunoglobulin isotypes, coupled with a pronounced reduction or absence of B-cells. Approximately 80 to 90% of individuals exhibit genetic variations in Bruton’s agammaglobulinemia tyrosine kinase (BTK), whereas a minority of cases, around 5–10%, are autosomal recessive agammaglobulinemia (ARA). Very few cases are grouped into distinct subcategories. We evaluated phenotypically and genetically 27 patients from 13 distinct families with hypogammaglobinemia and no B-cells. Genetic analysis was performed via whole-exome and Sanger sequencing. The most prevalent genetic cause was mutations in <i>BTK</i>. Three novel mutations in the <i>BTK</i> gene include c.115 T > C (p. Tyr39His), c.685-686insTTAC (p.Asn229llefs5), and c.163delT (p.Ser55GlnfsTer2). Our three ARA patients include a novel homozygous stop-gain mutation in the immunoglobulin heavy constant Mu chain (<i>IGHM</i>) gene, a novel frameshift mutation of the B-cell antigen receptor complex-associated protein (<i>CD79A</i>) gene, a novel bi-allelic stop-gain mutation in the transcription factor 3 (<i>TCF3</i>) gene. Three patients with agammaglobulinemia have an autosomal dominant inheritance pattern, which includes a missense variant in <i>PIK3CD</i>, a novel missense variant in <i>PIK3R1</i> and a homozygous silent mutation in the phosphoinositide-3-kinase regulatory subunit (<i>RASGRP1</i>) gene. This study broadens the genetic spectrum of hypogammaglobulinemia without B-cells and presented a few novel variants within the Iranian community, which may also have implications in other Middle Eastern populations. Notably, disease control was better in the second affected family member in families with multiple cases.</p>","PeriodicalId":13446,"journal":{"name":"Immunogenetics","volume":"73 1","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2024-04-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"B-cells absence in patients diagnosed as inborn errors of immunity: a registry-based study\",\"authors\":\"Razieh Khoshnevisan, Shakiba Hassanzadeh, Christoph Klein, Meino Rohlfs, Bodo Grimbacher, Newsha Molavi, Aryana Zamanifar, Ali Khoshnevisan, Mahbube Jafari, Bahram Bagherpour, Mahdiyeh Behnam, Somayeh Najafi, Roya Sherkat\",\"doi\":\"10.1007/s00251-024-01342-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Hypogammaglobulinemia without B-cells is a subgroup of inborn errors of immunity (IEI) which is characterized by a significant decline in all serum immunoglobulin isotypes, coupled with a pronounced reduction or absence of B-cells. Approximately 80 to 90% of individuals exhibit genetic variations in Bruton’s agammaglobulinemia tyrosine kinase (BTK), whereas a minority of cases, around 5–10%, are autosomal recessive agammaglobulinemia (ARA). Very few cases are grouped into distinct subcategories. We evaluated phenotypically and genetically 27 patients from 13 distinct families with hypogammaglobinemia and no B-cells. Genetic analysis was performed via whole-exome and Sanger sequencing. The most prevalent genetic cause was mutations in <i>BTK</i>. Three novel mutations in the <i>BTK</i> gene include c.115 T > C (p. Tyr39His), c.685-686insTTAC (p.Asn229llefs5), and c.163delT (p.Ser55GlnfsTer2). Our three ARA patients include a novel homozygous stop-gain mutation in the immunoglobulin heavy constant Mu chain (<i>IGHM</i>) gene, a novel frameshift mutation of the B-cell antigen receptor complex-associated protein (<i>CD79A</i>) gene, a novel bi-allelic stop-gain mutation in the transcription factor 3 (<i>TCF3</i>) gene. Three patients with agammaglobulinemia have an autosomal dominant inheritance pattern, which includes a missense variant in <i>PIK3CD</i>, a novel missense variant in <i>PIK3R1</i> and a homozygous silent mutation in the phosphoinositide-3-kinase regulatory subunit (<i>RASGRP1</i>) gene. This study broadens the genetic spectrum of hypogammaglobulinemia without B-cells and presented a few novel variants within the Iranian community, which may also have implications in other Middle Eastern populations. Notably, disease control was better in the second affected family member in families with multiple cases.</p>\",\"PeriodicalId\":13446,\"journal\":{\"name\":\"Immunogenetics\",\"volume\":\"73 1\",\"pages\":\"\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-04-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Immunogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s00251-024-01342-y\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00251-024-01342-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
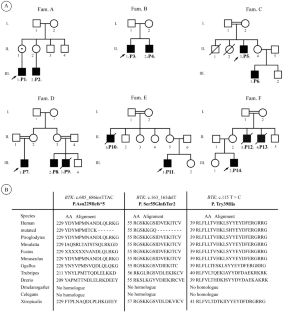
无 B 细胞的低丙种球蛋白血症是先天性免疫错误(IEI)的一个亚组,其特征是所有血清免疫球蛋白异型显著下降,同时 B 细胞明显减少或缺失。约 80% 至 90% 的患者表现出布鲁顿酪氨酸激酶(BTK)的遗传变异,而少数病例(约 5% 至 10%)为常染色体隐性遗传性丙种球蛋白血症(ARA)。只有极少数病例可分为不同的亚类。我们对来自 13 个不同家族的 27 名患有低丙种球蛋白血症且无 B 细胞的患者进行了表型和遗传评估。遗传分析是通过全外显子组和桑格测序法进行的。最常见的遗传原因是 BTK 基因突变。BTK 基因的三个新型突变包括 c.115 T > C(p.Tyr39His)、c.685-686insTTAC(p.Asn229llefs5)和 c.163delT(p.Ser55GlnfsTer2)。我们的 3 位 ARA 患者包括免疫球蛋白重常 Mu 链(IGHM)基因的新型同源终止-增益突变、B 细胞抗原受体复合物相关蛋白(CD79A)基因的新型换框突变、转录因子 3(TCF3)基因的新型双等位终止-增益突变。三位农夫球蛋白血症患者具有常染色体显性遗传模式,其中包括 PIK3CD 基因的一个错义变体、PIK3R1 基因的一个新型错义变体以及磷脂酰肌醇-3-激酶调节亚基(RASGRP1)基因的一个同源沉默突变。这项研究拓宽了无 B 细胞低丙种球蛋白血症的基因谱,并在伊朗人群体中发现了一些新型变异体,这可能对其他中东人群也有影响。值得注意的是,在有多个病例的家庭中,第二个受影响家庭成员的疾病控制情况更好。
B-cells absence in patients diagnosed as inborn errors of immunity: a registry-based study
Hypogammaglobulinemia without B-cells is a subgroup of inborn errors of immunity (IEI) which is characterized by a significant decline in all serum immunoglobulin isotypes, coupled with a pronounced reduction or absence of B-cells. Approximately 80 to 90% of individuals exhibit genetic variations in Bruton’s agammaglobulinemia tyrosine kinase (BTK), whereas a minority of cases, around 5–10%, are autosomal recessive agammaglobulinemia (ARA). Very few cases are grouped into distinct subcategories. We evaluated phenotypically and genetically 27 patients from 13 distinct families with hypogammaglobinemia and no B-cells. Genetic analysis was performed via whole-exome and Sanger sequencing. The most prevalent genetic cause was mutations in BTK. Three novel mutations in the BTK gene include c.115 T > C (p. Tyr39His), c.685-686insTTAC (p.Asn229llefs5), and c.163delT (p.Ser55GlnfsTer2). Our three ARA patients include a novel homozygous stop-gain mutation in the immunoglobulin heavy constant Mu chain (IGHM) gene, a novel frameshift mutation of the B-cell antigen receptor complex-associated protein (CD79A) gene, a novel bi-allelic stop-gain mutation in the transcription factor 3 (TCF3) gene. Three patients with agammaglobulinemia have an autosomal dominant inheritance pattern, which includes a missense variant in PIK3CD, a novel missense variant in PIK3R1 and a homozygous silent mutation in the phosphoinositide-3-kinase regulatory subunit (RASGRP1) gene. This study broadens the genetic spectrum of hypogammaglobulinemia without B-cells and presented a few novel variants within the Iranian community, which may also have implications in other Middle Eastern populations. Notably, disease control was better in the second affected family member in families with multiple cases.
期刊介绍:
Immunogenetics publishes original papers, brief communications, and reviews on research in the following areas: genetics and evolution of the immune system; genetic control of immune response and disease susceptibility; bioinformatics of the immune system; structure of immunologically important molecules; and immunogenetics of reproductive biology, tissue differentiation, and development.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
