{"title":"利用支持向量机将结肠癌患者分为一致认可的分子亚型。","authors":"Necla Koçhan, Barış Emre Dayanç","doi":"10.55730/1300-0152.2674","DOIUrl":null,"url":null,"abstract":"<p><strong>Background/aim: </strong>The molecular heterogeneity of colon cancer has made classification of tumors a requirement for effective treatment. One of the approaches for molecular subtyping of colon cancer patients is the consensus molecular subtypes (CMS), developed by the Colorectal Cancer Subtyping Consortium. CMS-specific RNA-Seq-dependent classification approaches are recent, with relatively low sensitivity and specificity. In this study, we aimed to classify patients into CMS groups using their RNA-seq profiles.</p><p><strong>Materials and methods: </strong>We first identified subtype-specific and survival-associated genes using the Fuzzy C-Means algorithm and log-rank test. We then classified patients using support vector machines with backward elimination methodology.</p><p><strong>Results: </strong>We optimized RNA-seq-based classification using 25 genes with a minimum classification error rate. In this study, we reported the classification performance using precision, sensitivity, specificity, false discovery rate, and balanced accuracy metrics.</p><p><strong>Conclusion: </strong>We present a gene list for colon cancer classification with minimum classification error rates and observed the lowest sensitivity but the highest specificity with CMS3-associated genes, which significantly differed due to the low number of patients in the clinic for this group.</p>","PeriodicalId":94363,"journal":{"name":"Turkish journal of biology = Turk biyoloji dergisi","volume":"47 6","pages":"406-412"},"PeriodicalIF":0.9000,"publicationDate":"2023-12-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11045208/pdf/","citationCount":"0","resultStr":"{\"title\":\"Classification of colon cancer patients into consensus molecular subtypes using support vector machines.\",\"authors\":\"Necla Koçhan, Barış Emre Dayanç\",\"doi\":\"10.55730/1300-0152.2674\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background/aim: </strong>The molecular heterogeneity of colon cancer has made classification of tumors a requirement for effective treatment. One of the approaches for molecular subtyping of colon cancer patients is the consensus molecular subtypes (CMS), developed by the Colorectal Cancer Subtyping Consortium. CMS-specific RNA-Seq-dependent classification approaches are recent, with relatively low sensitivity and specificity. In this study, we aimed to classify patients into CMS groups using their RNA-seq profiles.</p><p><strong>Materials and methods: </strong>We first identified subtype-specific and survival-associated genes using the Fuzzy C-Means algorithm and log-rank test. We then classified patients using support vector machines with backward elimination methodology.</p><p><strong>Results: </strong>We optimized RNA-seq-based classification using 25 genes with a minimum classification error rate. In this study, we reported the classification performance using precision, sensitivity, specificity, false discovery rate, and balanced accuracy metrics.</p><p><strong>Conclusion: </strong>We present a gene list for colon cancer classification with minimum classification error rates and observed the lowest sensitivity but the highest specificity with CMS3-associated genes, which significantly differed due to the low number of patients in the clinic for this group.</p>\",\"PeriodicalId\":94363,\"journal\":{\"name\":\"Turkish journal of biology = Turk biyoloji dergisi\",\"volume\":\"47 6\",\"pages\":\"406-412\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2023-12-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11045208/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Turkish journal of biology = Turk biyoloji dergisi\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.55730/1300-0152.2674\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Turkish journal of biology = Turk biyoloji dergisi","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.55730/1300-0152.2674","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Classification of colon cancer patients into consensus molecular subtypes using support vector machines.
Background/aim: The molecular heterogeneity of colon cancer has made classification of tumors a requirement for effective treatment. One of the approaches for molecular subtyping of colon cancer patients is the consensus molecular subtypes (CMS), developed by the Colorectal Cancer Subtyping Consortium. CMS-specific RNA-Seq-dependent classification approaches are recent, with relatively low sensitivity and specificity. In this study, we aimed to classify patients into CMS groups using their RNA-seq profiles.
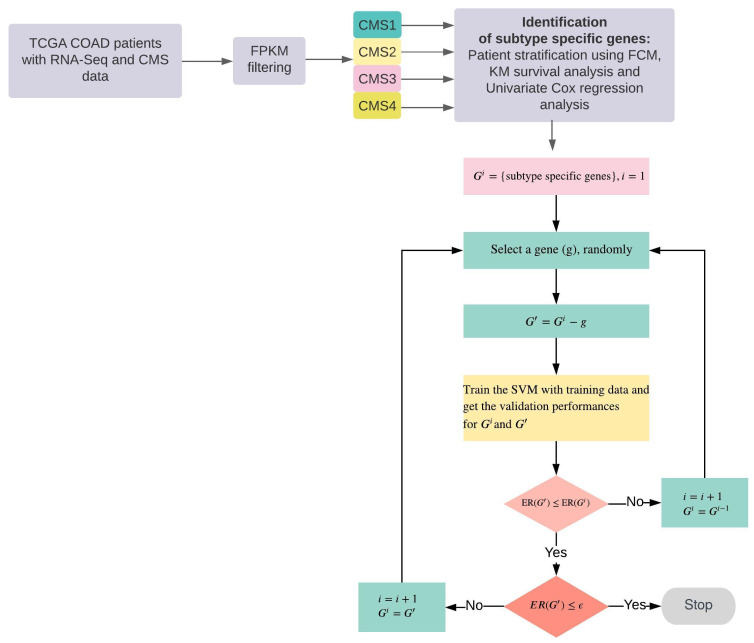
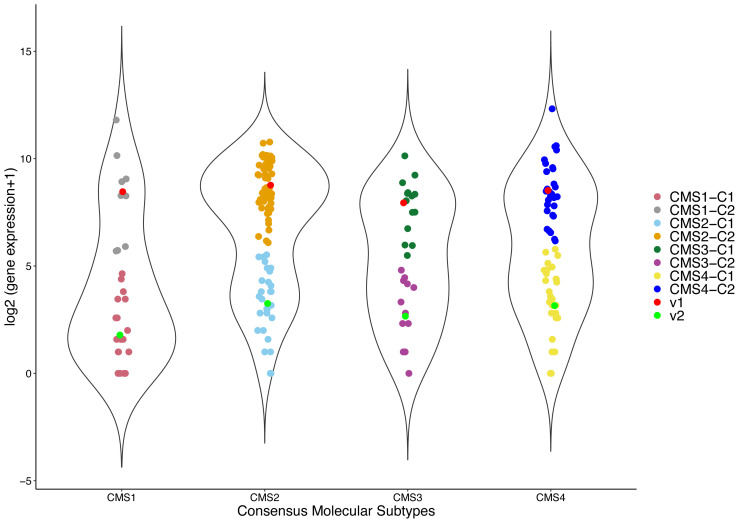
Materials and methods: We first identified subtype-specific and survival-associated genes using the Fuzzy C-Means algorithm and log-rank test. We then classified patients using support vector machines with backward elimination methodology.
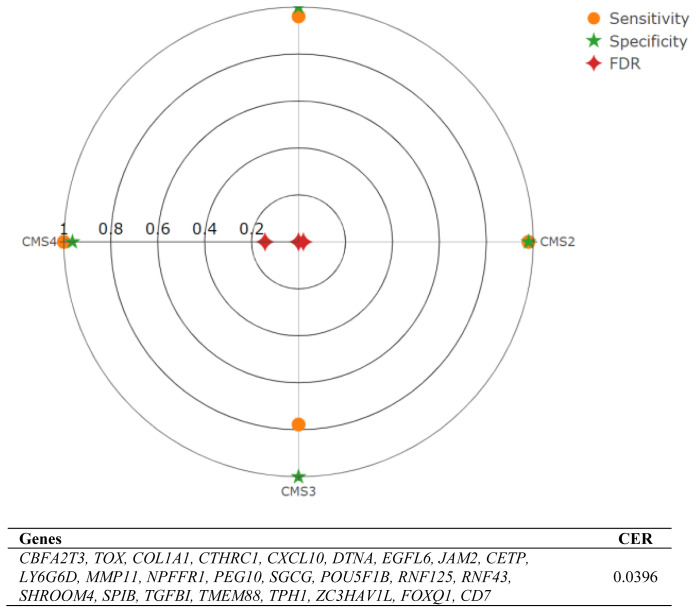
Results: We optimized RNA-seq-based classification using 25 genes with a minimum classification error rate. In this study, we reported the classification performance using precision, sensitivity, specificity, false discovery rate, and balanced accuracy metrics.
Conclusion: We present a gene list for colon cancer classification with minimum classification error rates and observed the lowest sensitivity but the highest specificity with CMS3-associated genes, which significantly differed due to the low number of patients in the clinic for this group.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
