Jun Takayama, Satoshi Makino, Takamitsu Funayama, Masao Ueki, Akira Narita, Keiko Murakami, Masatsugu Orui, Mami Ishikuro, Taku Obara, the Tohoku Medical Megabank Project Study Group, Shinichi Kuriyama, Masayuki Yamamoto, Gen Tamiya
{"title":"日本人口的精细遗传图谱。","authors":"Jun Takayama, Satoshi Makino, Takamitsu Funayama, Masao Ueki, Akira Narita, Keiko Murakami, Masatsugu Orui, Mami Ishikuro, Taku Obara, the Tohoku Medical Megabank Project Study Group, Shinichi Kuriyama, Masayuki Yamamoto, Gen Tamiya","doi":"10.1111/cge.14536","DOIUrl":null,"url":null,"abstract":"<p>Genetic maps are fundamental resources for linkage and association studies. A fine-scale genetic map can be constructed by inferring historical recombination events from the genome-wide structure of linkage disequilibrium—a non-random association of alleles among loci—by using population-scale sequencing data. We constructed a fine-scale genetic map and identified recombination hotspots from 10 092 551 bi-allelic high-quality autosomal markers segregating among 150 unrelated Japanese individuals whose genotypes were determined by high-coverage (30×) whole-genome sequencing, and the genotype quality was carefully controlled by using their parents' and offspring's genotypes. The pedigree information was also utilized for haplotype phasing. The resulting genome-wide recombination rate profiles were concordant with those of the worldwide population on a broad scale, and the resolution was much improved. We identified 9487 recombination hotspots and confirmed the enrichment of previously known motifs in the hotspots. Moreover, we demonstrated that the Japanese genetic map improved the haplotype phasing and genotype imputation accuracy for the Japanese population. The construction of a population-specific genetic map will help make genetics research more accurate.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":"106 3","pages":"284-292"},"PeriodicalIF":2.3000,"publicationDate":"2024-05-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14536","citationCount":"0","resultStr":"{\"title\":\"A fine-scale genetic map of the Japanese population\",\"authors\":\"Jun Takayama, Satoshi Makino, Takamitsu Funayama, Masao Ueki, Akira Narita, Keiko Murakami, Masatsugu Orui, Mami Ishikuro, Taku Obara, the Tohoku Medical Megabank Project Study Group, Shinichi Kuriyama, Masayuki Yamamoto, Gen Tamiya\",\"doi\":\"10.1111/cge.14536\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Genetic maps are fundamental resources for linkage and association studies. A fine-scale genetic map can be constructed by inferring historical recombination events from the genome-wide structure of linkage disequilibrium—a non-random association of alleles among loci—by using population-scale sequencing data. We constructed a fine-scale genetic map and identified recombination hotspots from 10 092 551 bi-allelic high-quality autosomal markers segregating among 150 unrelated Japanese individuals whose genotypes were determined by high-coverage (30×) whole-genome sequencing, and the genotype quality was carefully controlled by using their parents' and offspring's genotypes. The pedigree information was also utilized for haplotype phasing. The resulting genome-wide recombination rate profiles were concordant with those of the worldwide population on a broad scale, and the resolution was much improved. We identified 9487 recombination hotspots and confirmed the enrichment of previously known motifs in the hotspots. Moreover, we demonstrated that the Japanese genetic map improved the haplotype phasing and genotype imputation accuracy for the Japanese population. The construction of a population-specific genetic map will help make genetics research more accurate.</p>\",\"PeriodicalId\":10354,\"journal\":{\"name\":\"Clinical Genetics\",\"volume\":\"106 3\",\"pages\":\"284-292\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2024-05-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14536\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cge.14536\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14536","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
A fine-scale genetic map of the Japanese population
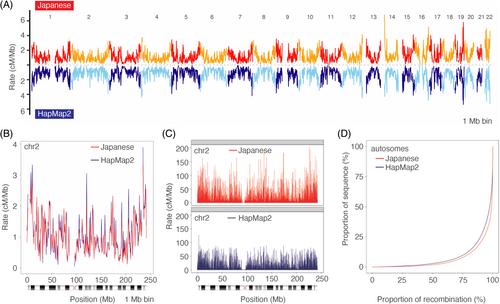
Genetic maps are fundamental resources for linkage and association studies. A fine-scale genetic map can be constructed by inferring historical recombination events from the genome-wide structure of linkage disequilibrium—a non-random association of alleles among loci—by using population-scale sequencing data. We constructed a fine-scale genetic map and identified recombination hotspots from 10 092 551 bi-allelic high-quality autosomal markers segregating among 150 unrelated Japanese individuals whose genotypes were determined by high-coverage (30×) whole-genome sequencing, and the genotype quality was carefully controlled by using their parents' and offspring's genotypes. The pedigree information was also utilized for haplotype phasing. The resulting genome-wide recombination rate profiles were concordant with those of the worldwide population on a broad scale, and the resolution was much improved. We identified 9487 recombination hotspots and confirmed the enrichment of previously known motifs in the hotspots. Moreover, we demonstrated that the Japanese genetic map improved the haplotype phasing and genotype imputation accuracy for the Japanese population. The construction of a population-specific genetic map will help make genetics research more accurate.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
