Liliya R. Safina , Elizaveta A. Rozhnova , Karina A. Krylova , Ramil T. Murzaev , Julia A. Baimova
{"title":"石墨烯增强金属复合材料的原子间势垒:最佳选择","authors":"Liliya R. Safina , Elizaveta A. Rozhnova , Karina A. Krylova , Ramil T. Murzaev , Julia A. Baimova","doi":"10.1016/j.cpc.2024.109235","DOIUrl":null,"url":null,"abstract":"<div><p>Graphene reinforced metal matrix composites represent a promising class of materials for high-strength surface coatings because of their high strength and ductility. This study reports the application of different interatomic potentials to correctly describe the interaction between graphene and metals (Al, Cu, Ni, and Ti) by molecular dynamics. Both simple pair potentials, such as Lennard-Jones and Morse, and many-body potentials, such as bond order potential are applied for the simulation of a graphene/metal system at room temperature. Three different structures are considered: (i) graphene interacting with one metal atom; (ii) graphene interacting with a metal nanoparticle, and (iii) three-dimensional graphene network filled with metal nanoparticles. We first determine the potential energy that any graphene/metal system can reach during exposure at 300 K; then, we analyze the interaction dynamics for all considered systems and all potentials. A considerable difference in the interaction between metal nanoparticles with planar and folded graphene was found. For graphene/Ni, graphene/Cu, and graphene/Ti, the Lennard-Jones and Morse potentials provide accurate energetic and structural properties of the studied structures; they also describe interaction in the graphene/metal system in a similar way, at variance with bond-order potential. For graphene/Al, the Tersoff and Morse potentials describe the interaction better than Lennard-Jones. For the simulation of graphene/Me system, the optimal choice of the potential for different structures is of crucial importance. The presented analysis of the interatomic potentials appears to be promising for realistic and accurate simulations of graphene reinforced metal composites.</p></div>","PeriodicalId":285,"journal":{"name":"Computer Physics Communications","volume":"301 ","pages":"Article 109235"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Interatomic potentials for graphene reinforced metal composites: Optimal choice\",\"authors\":\"Liliya R. Safina , Elizaveta A. Rozhnova , Karina A. Krylova , Ramil T. Murzaev , Julia A. Baimova\",\"doi\":\"10.1016/j.cpc.2024.109235\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Graphene reinforced metal matrix composites represent a promising class of materials for high-strength surface coatings because of their high strength and ductility. This study reports the application of different interatomic potentials to correctly describe the interaction between graphene and metals (Al, Cu, Ni, and Ti) by molecular dynamics. Both simple pair potentials, such as Lennard-Jones and Morse, and many-body potentials, such as bond order potential are applied for the simulation of a graphene/metal system at room temperature. Three different structures are considered: (i) graphene interacting with one metal atom; (ii) graphene interacting with a metal nanoparticle, and (iii) three-dimensional graphene network filled with metal nanoparticles. We first determine the potential energy that any graphene/metal system can reach during exposure at 300 K; then, we analyze the interaction dynamics for all considered systems and all potentials. A considerable difference in the interaction between metal nanoparticles with planar and folded graphene was found. For graphene/Ni, graphene/Cu, and graphene/Ti, the Lennard-Jones and Morse potentials provide accurate energetic and structural properties of the studied structures; they also describe interaction in the graphene/metal system in a similar way, at variance with bond-order potential. For graphene/Al, the Tersoff and Morse potentials describe the interaction better than Lennard-Jones. For the simulation of graphene/Me system, the optimal choice of the potential for different structures is of crucial importance. The presented analysis of the interatomic potentials appears to be promising for realistic and accurate simulations of graphene reinforced metal composites.</p></div>\",\"PeriodicalId\":285,\"journal\":{\"name\":\"Computer Physics Communications\",\"volume\":\"301 \",\"pages\":\"Article 109235\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computer Physics Communications\",\"FirstCategoryId\":\"101\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0010465524001589\",\"RegionNum\":2,\"RegionCategory\":\"物理与天体物理\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/5/8 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computer Physics Communications","FirstCategoryId":"101","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0010465524001589","RegionNum":2,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/8 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
引用次数: 0
摘要
石墨烯增强金属基复合材料具有高强度和延展性,是一类很有前途的高强度表面涂层材料。本研究报告了不同原子间势的应用,以通过分子动力学正确描述石墨烯与金属(铝、铜、镍和钛)之间的相互作用。在模拟室温下的石墨烯/金属体系时,既应用了简单的对势能(如伦纳德-琼斯和莫尔斯),也应用了多体势能(如键阶势能)。我们考虑了三种不同的结构:(i) 与一个金属原子相互作用的石墨烯;(ii) 与一个金属纳米粒子相互作用的石墨烯;(iii) 充满金属纳米粒子的三维石墨烯网络。我们首先确定了任何石墨烯/金属系统在 300 K 暴露条件下可达到的势能,然后分析了所有考虑的系统和所有势能的相互作用动力学。我们发现金属纳米粒子与平面石墨烯和折叠石墨烯之间的相互作用存在很大差异。对于石墨烯/尼、石墨烯/铜和石墨烯/钛,伦纳德-琼斯电位和莫尔斯电位提供了所研究结构的精确能量和结构特性;它们也以类似的方式描述了石墨烯/金属体系中的相互作用,但与键阶电位不同。对于石墨烯/铝,特尔索夫势和莫尔斯势比伦纳德-琼斯势更好地描述了相互作用。对于石墨烯/Me 体系的模拟,不同结构的最佳电位选择至关重要。本文介绍的原子间电位分析似乎有望实现对石墨烯增强金属复合材料的真实、准确模拟。
Interatomic potentials for graphene reinforced metal composites: Optimal choice
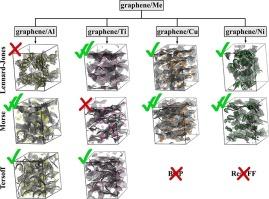
Graphene reinforced metal matrix composites represent a promising class of materials for high-strength surface coatings because of their high strength and ductility. This study reports the application of different interatomic potentials to correctly describe the interaction between graphene and metals (Al, Cu, Ni, and Ti) by molecular dynamics. Both simple pair potentials, such as Lennard-Jones and Morse, and many-body potentials, such as bond order potential are applied for the simulation of a graphene/metal system at room temperature. Three different structures are considered: (i) graphene interacting with one metal atom; (ii) graphene interacting with a metal nanoparticle, and (iii) three-dimensional graphene network filled with metal nanoparticles. We first determine the potential energy that any graphene/metal system can reach during exposure at 300 K; then, we analyze the interaction dynamics for all considered systems and all potentials. A considerable difference in the interaction between metal nanoparticles with planar and folded graphene was found. For graphene/Ni, graphene/Cu, and graphene/Ti, the Lennard-Jones and Morse potentials provide accurate energetic and structural properties of the studied structures; they also describe interaction in the graphene/metal system in a similar way, at variance with bond-order potential. For graphene/Al, the Tersoff and Morse potentials describe the interaction better than Lennard-Jones. For the simulation of graphene/Me system, the optimal choice of the potential for different structures is of crucial importance. The presented analysis of the interatomic potentials appears to be promising for realistic and accurate simulations of graphene reinforced metal composites.
期刊介绍:
The focus of CPC is on contemporary computational methods and techniques and their implementation, the effectiveness of which will normally be evidenced by the author(s) within the context of a substantive problem in physics. Within this setting CPC publishes two types of paper.
Computer Programs in Physics (CPiP)
These papers describe significant computer programs to be archived in the CPC Program Library which is held in the Mendeley Data repository. The submitted software must be covered by an approved open source licence. Papers and associated computer programs that address a problem of contemporary interest in physics that cannot be solved by current software are particularly encouraged.
Computational Physics Papers (CP)
These are research papers in, but are not limited to, the following themes across computational physics and related disciplines.
mathematical and numerical methods and algorithms;
computational models including those associated with the design, control and analysis of experiments; and
algebraic computation.
Each will normally include software implementation and performance details. The software implementation should, ideally, be available via GitHub, Zenodo or an institutional repository.In addition, research papers on the impact of advanced computer architecture and special purpose computers on computing in the physical sciences and software topics related to, and of importance in, the physical sciences may be considered.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
