Anni Saarela, Oskari Timonen, Jarkko Kirjavainen, Yawu Liu, Katri Silvennoinen, Esa Mervaala, Reetta Kälviäinen
{"title":"芬兰人群中富集的新型 LAMC3 致病变体会导致大脑皮层发育畸形和严重癫痫。","authors":"Anni Saarela, Oskari Timonen, Jarkko Kirjavainen, Yawu Liu, Katri Silvennoinen, Esa Mervaala, Reetta Kälviäinen","doi":"10.1002/epd2.20244","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Objective</h3>\n \n <p>Recessive <i>LAMC3</i> mutations are recognized to cause epilepsy with cortical malformations characterized by polymicrogyria and pachygyria. The objective of this study was to describe the clinical picture and epilepsy phenotype of four patients with a previously undescribed <i>LAMC3</i> variant.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>All epilepsy patients treated in Kuopio Epilepsy Center (located in Kuopio, Finland) are offered the possibility to participate in a scientific study investigating biomarkers in epilepsy (Epibiomarker study). We have collected a comprehensive database of the study population, and are currently re-evaluating our database regarding the patients with developmental and/or epileptic encephalopathy (DEE). If the etiology of epilepsy remains unknown in the clinical setting, we are performing whole exome sequencing to recognize the genetic causes.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Among our study population of 323 DEE patients we recognized three patients with similar homozygous <i>LAMC3</i> c.1866del (p.(Phe623Serfs*10)) frameshift variant and one patient with a compound heterozygous mutation where the same frameshift variant was combined with an intronic <i>LAMC3</i> c.4231-12C>G variant on another allele. All these patients have severe epilepsy and either bilateral agyria-pachygyria or bilateral polymicrogyria in their clinical MRI scanning. Cortical malformations involve the occipital lobes in all our patients. Epilepsy phenotype is variable as two of our patients have DEE with epileptic spasms progressing to Lennox–Gastaut syndrome and intellectual disability. The other two patients have focal epilepsy without marked cognitive deficit. The four patients are unrelated. <i>LAMC3</i> c.1866del p.(Phe623Serfs*10) frameshift variant is enriched in the Finnish population.</p>\n </section>\n \n <section>\n \n <h3> Significance</h3>\n \n <p>Only a few patients with epilepsy caused by <i>LAMC3</i> homozygous or compound heterozygous mutations have been described in the literature. To our knowledge, the variants discovered in our patients have not previously been published. Clinical phenotype appears to be more varied than previously assumed and patients with a milder phenotype and normal cognition have probably remained unrecognized.</p>\n </section>\n </div>","PeriodicalId":50508,"journal":{"name":"Epileptic Disorders","volume":"26 4","pages":"498-509"},"PeriodicalIF":2.7000,"publicationDate":"2024-05-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/epd2.20244","citationCount":"0","resultStr":"{\"title\":\"Novel LAMC3 pathogenic variant enriched in Finnish population causes malformations of cortical development and severe epilepsy\",\"authors\":\"Anni Saarela, Oskari Timonen, Jarkko Kirjavainen, Yawu Liu, Katri Silvennoinen, Esa Mervaala, Reetta Kälviäinen\",\"doi\":\"10.1002/epd2.20244\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Objective</h3>\\n \\n <p>Recessive <i>LAMC3</i> mutations are recognized to cause epilepsy with cortical malformations characterized by polymicrogyria and pachygyria. The objective of this study was to describe the clinical picture and epilepsy phenotype of four patients with a previously undescribed <i>LAMC3</i> variant.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>All epilepsy patients treated in Kuopio Epilepsy Center (located in Kuopio, Finland) are offered the possibility to participate in a scientific study investigating biomarkers in epilepsy (Epibiomarker study). We have collected a comprehensive database of the study population, and are currently re-evaluating our database regarding the patients with developmental and/or epileptic encephalopathy (DEE). If the etiology of epilepsy remains unknown in the clinical setting, we are performing whole exome sequencing to recognize the genetic causes.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Among our study population of 323 DEE patients we recognized three patients with similar homozygous <i>LAMC3</i> c.1866del (p.(Phe623Serfs*10)) frameshift variant and one patient with a compound heterozygous mutation where the same frameshift variant was combined with an intronic <i>LAMC3</i> c.4231-12C>G variant on another allele. All these patients have severe epilepsy and either bilateral agyria-pachygyria or bilateral polymicrogyria in their clinical MRI scanning. Cortical malformations involve the occipital lobes in all our patients. Epilepsy phenotype is variable as two of our patients have DEE with epileptic spasms progressing to Lennox–Gastaut syndrome and intellectual disability. The other two patients have focal epilepsy without marked cognitive deficit. The four patients are unrelated. <i>LAMC3</i> c.1866del p.(Phe623Serfs*10) frameshift variant is enriched in the Finnish population.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Significance</h3>\\n \\n <p>Only a few patients with epilepsy caused by <i>LAMC3</i> homozygous or compound heterozygous mutations have been described in the literature. To our knowledge, the variants discovered in our patients have not previously been published. Clinical phenotype appears to be more varied than previously assumed and patients with a milder phenotype and normal cognition have probably remained unrecognized.</p>\\n </section>\\n </div>\",\"PeriodicalId\":50508,\"journal\":{\"name\":\"Epileptic Disorders\",\"volume\":\"26 4\",\"pages\":\"498-509\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-05-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/epd2.20244\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Epileptic Disorders\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/epd2.20244\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epileptic Disorders","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/epd2.20244","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Novel LAMC3 pathogenic variant enriched in Finnish population causes malformations of cortical development and severe epilepsy
Objective
Recessive LAMC3 mutations are recognized to cause epilepsy with cortical malformations characterized by polymicrogyria and pachygyria. The objective of this study was to describe the clinical picture and epilepsy phenotype of four patients with a previously undescribed LAMC3 variant.
Methods
All epilepsy patients treated in Kuopio Epilepsy Center (located in Kuopio, Finland) are offered the possibility to participate in a scientific study investigating biomarkers in epilepsy (Epibiomarker study). We have collected a comprehensive database of the study population, and are currently re-evaluating our database regarding the patients with developmental and/or epileptic encephalopathy (DEE). If the etiology of epilepsy remains unknown in the clinical setting, we are performing whole exome sequencing to recognize the genetic causes.
Results
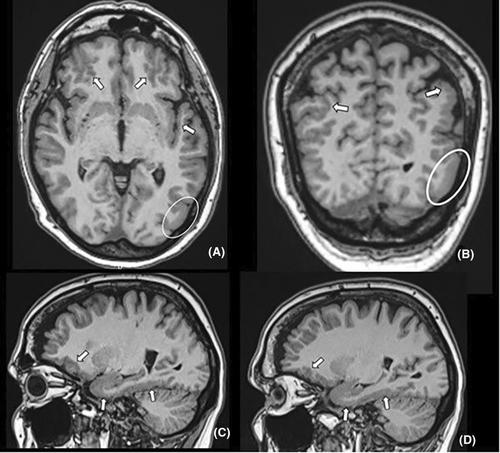
Among our study population of 323 DEE patients we recognized three patients with similar homozygous LAMC3 c.1866del (p.(Phe623Serfs*10)) frameshift variant and one patient with a compound heterozygous mutation where the same frameshift variant was combined with an intronic LAMC3 c.4231-12C>G variant on another allele. All these patients have severe epilepsy and either bilateral agyria-pachygyria or bilateral polymicrogyria in their clinical MRI scanning. Cortical malformations involve the occipital lobes in all our patients. Epilepsy phenotype is variable as two of our patients have DEE with epileptic spasms progressing to Lennox–Gastaut syndrome and intellectual disability. The other two patients have focal epilepsy without marked cognitive deficit. The four patients are unrelated. LAMC3 c.1866del p.(Phe623Serfs*10) frameshift variant is enriched in the Finnish population.
Significance
Only a few patients with epilepsy caused by LAMC3 homozygous or compound heterozygous mutations have been described in the literature. To our knowledge, the variants discovered in our patients have not previously been published. Clinical phenotype appears to be more varied than previously assumed and patients with a milder phenotype and normal cognition have probably remained unrecognized.
期刊介绍:
Epileptic Disorders is the leading forum where all experts and medical studentswho wish to improve their understanding of epilepsy and related disorders can share practical experiences surrounding diagnosis and care, natural history, and management of seizures.
Epileptic Disorders is the official E-journal of the International League Against Epilepsy for educational communication. As the journal celebrates its 20th anniversary, it will now be available only as an online version. Its mission is to create educational links between epileptologists and other health professionals in clinical practice and scientists or physicians in research-based institutions. This change is accompanied by an increase in the number of issues per year, from 4 to 6, to ensure regular diffusion of recently published material (high quality Review and Seminar in Epileptology papers; Original Research articles or Case reports of educational value; MultiMedia Teaching Material), to serve the global medical community that cares for those affected by epilepsy.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
