Carolin Joy, Bikramaditya Mandal, Dulat Bostan, Marie-Lise Dubernet and Dmitri Babikov
{"title":"复杂系统中分子-分子碰撞动力学的量子/经典混合理论(MQCT)方法。","authors":"Carolin Joy, Bikramaditya Mandal, Dulat Bostan, Marie-Lise Dubernet and Dmitri Babikov","doi":"10.1039/D3FD00166K","DOIUrl":null,"url":null,"abstract":"<p >We developed a general theoretical approach and a user-ready computer code that permit study of the dynamics of collisional energy transfer and ro-vibrational energy exchange in complex molecule–molecule collisions. The method is a mixture of classical and quantum mechanics. The internal ro-vibrational motion of collision partners is treated quantum mechanically using a time-dependent Schrödinger equation that captures many quantum phenomena including state quantization and zero-point energy, propensity and selection rules for state-to-state transitions, quantum symmetry and interference phenomena. A significant numerical speed up is obtained by describing the translational motion of collision partners classically, using the Ehrenfest mean-field trajectory approach. Within this framework a family of approximate methods for collision dynamics is developed. Several benchmark studies for diatomic and triatomic molecules, such as H<small><sub>2</sub></small>O and ND<small><sub>3</sub></small> collided with He, H<small><sub>2</sub></small> and D<small><sub>2</sub></small>, show that the results of MQCT are in good agreement with full-quantum calculations in a broad range of energies, especially at high collision energies where they become nearly identical to the full quantum results. Numerical efficiency of the method and massive parallelism of the MQCT code permit us to embrace some of the most complicated collisional systems ever studied, such as C<small><sub>6</sub></small>H<small><sub>6</sub></small> + He, CH<small><sub>3</sub></small>COOH + He and H<small><sub>2</sub></small>O + H<small><sub>2</sub></small>O. Application of MQCT to the collisions of chiral molecules such as CH<small><sub>3</sub></small>CHCH<small><sub>2</sub></small>O + He, and to molecule–surface collisions is also possible and will be pursued in the future.</p>","PeriodicalId":49075,"journal":{"name":"Faraday Discussions","volume":"251 ","pages":" 225-248"},"PeriodicalIF":3.1000,"publicationDate":"2024-05-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Mixed quantum/classical theory (MQCT) approach to the dynamics of molecule–molecule collisions in complex systems†\",\"authors\":\"Carolin Joy, Bikramaditya Mandal, Dulat Bostan, Marie-Lise Dubernet and Dmitri Babikov\",\"doi\":\"10.1039/D3FD00166K\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We developed a general theoretical approach and a user-ready computer code that permit study of the dynamics of collisional energy transfer and ro-vibrational energy exchange in complex molecule–molecule collisions. The method is a mixture of classical and quantum mechanics. The internal ro-vibrational motion of collision partners is treated quantum mechanically using a time-dependent Schrödinger equation that captures many quantum phenomena including state quantization and zero-point energy, propensity and selection rules for state-to-state transitions, quantum symmetry and interference phenomena. A significant numerical speed up is obtained by describing the translational motion of collision partners classically, using the Ehrenfest mean-field trajectory approach. Within this framework a family of approximate methods for collision dynamics is developed. Several benchmark studies for diatomic and triatomic molecules, such as H<small><sub>2</sub></small>O and ND<small><sub>3</sub></small> collided with He, H<small><sub>2</sub></small> and D<small><sub>2</sub></small>, show that the results of MQCT are in good agreement with full-quantum calculations in a broad range of energies, especially at high collision energies where they become nearly identical to the full quantum results. Numerical efficiency of the method and massive parallelism of the MQCT code permit us to embrace some of the most complicated collisional systems ever studied, such as C<small><sub>6</sub></small>H<small><sub>6</sub></small> + He, CH<small><sub>3</sub></small>COOH + He and H<small><sub>2</sub></small>O + H<small><sub>2</sub></small>O. Application of MQCT to the collisions of chiral molecules such as CH<small><sub>3</sub></small>CHCH<small><sub>2</sub></small>O + He, and to molecule–surface collisions is also possible and will be pursued in the future.</p>\",\"PeriodicalId\":49075,\"journal\":{\"name\":\"Faraday Discussions\",\"volume\":\"251 \",\"pages\":\" 225-248\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2024-05-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Faraday Discussions\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d3fd00166k\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Faraday Discussions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d3fd00166k","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Chemistry","Score":null,"Total":0}
Mixed quantum/classical theory (MQCT) approach to the dynamics of molecule–molecule collisions in complex systems†
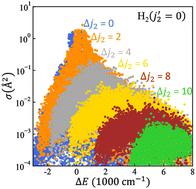
We developed a general theoretical approach and a user-ready computer code that permit study of the dynamics of collisional energy transfer and ro-vibrational energy exchange in complex molecule–molecule collisions. The method is a mixture of classical and quantum mechanics. The internal ro-vibrational motion of collision partners is treated quantum mechanically using a time-dependent Schrödinger equation that captures many quantum phenomena including state quantization and zero-point energy, propensity and selection rules for state-to-state transitions, quantum symmetry and interference phenomena. A significant numerical speed up is obtained by describing the translational motion of collision partners classically, using the Ehrenfest mean-field trajectory approach. Within this framework a family of approximate methods for collision dynamics is developed. Several benchmark studies for diatomic and triatomic molecules, such as H2O and ND3 collided with He, H2 and D2, show that the results of MQCT are in good agreement with full-quantum calculations in a broad range of energies, especially at high collision energies where they become nearly identical to the full quantum results. Numerical efficiency of the method and massive parallelism of the MQCT code permit us to embrace some of the most complicated collisional systems ever studied, such as C6H6 + He, CH3COOH + He and H2O + H2O. Application of MQCT to the collisions of chiral molecules such as CH3CHCH2O + He, and to molecule–surface collisions is also possible and will be pursued in the future.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
