Elena J. Tucker, Michael F. Sharp, Anna Lokchine, Katrina M. Bell, Catherine S. Palmer, Brianna L. Kline, Gorjana Robevska, Jocelyn van den Bergen, Jérôme Dulon, Diana Stojanovski, Katie L. Ayers, Philippe Touraine, Wayne Crismani, Sylvie Jaillard, Andrew H. Sinclair
{"title":"在患有孤立性卵巢早衰的姐妹中检测到 FANCA 双叶变体。","authors":"Elena J. Tucker, Michael F. Sharp, Anna Lokchine, Katrina M. Bell, Catherine S. Palmer, Brianna L. Kline, Gorjana Robevska, Jocelyn van den Bergen, Jérôme Dulon, Diana Stojanovski, Katie L. Ayers, Philippe Touraine, Wayne Crismani, Sylvie Jaillard, Andrew H. Sinclair","doi":"10.1111/cge.14543","DOIUrl":null,"url":null,"abstract":"<p>Premature ovarian insufficiency is a common form of female infertility affecting up to 4% of women and characterised by amenorrhea with elevated gonadotropin before the age of 40. Oocytes require controlled DNA breakage and repair for homologous recombination and the maintenance of oocyte integrity. Biallelic disruption of the DNA damage repair gene, <i>Fanconi anemia complementation group A</i> (<i>FANCA)</i>, is a common cause of Fanconi anaemia, a syndrome characterised by bone marrow failure, cancer predisposition, physical anomalies and POI. There is ongoing dispute about the role of heterozygous <i>FANCA</i> variants in POI pathogenesis, with insufficient evidence supporting causation. Here, we have identified biallelic <i>FANCA</i> variants in French sisters presenting with POI, including a novel missense variant of uncertain significance and a likely pathogenic deletion that initially evaded detection. Functional studies indicated no discernible effect on DNA damage sensitivity in patient lymphoblasts. These novel <i>FANCA</i> variants add evidence that heterozygous loss of one allele is insufficient to cause DNA damage sensitivity and POI. We propose that intragenic deletions, that are relatively common in <i>FANCA</i>, may be missed without careful analysis, and could explain the presumed causation of heterozygous variants. Accurate variant curation is critical to optimise patient care and outcomes.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":"106 3","pages":"321-335"},"PeriodicalIF":2.1000,"publicationDate":"2024-05-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14543","citationCount":"0","resultStr":"{\"title\":\"Biallelic FANCA variants detected in sisters with isolated premature ovarian insufficiency\",\"authors\":\"Elena J. Tucker, Michael F. Sharp, Anna Lokchine, Katrina M. Bell, Catherine S. Palmer, Brianna L. Kline, Gorjana Robevska, Jocelyn van den Bergen, Jérôme Dulon, Diana Stojanovski, Katie L. Ayers, Philippe Touraine, Wayne Crismani, Sylvie Jaillard, Andrew H. Sinclair\",\"doi\":\"10.1111/cge.14543\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Premature ovarian insufficiency is a common form of female infertility affecting up to 4% of women and characterised by amenorrhea with elevated gonadotropin before the age of 40. Oocytes require controlled DNA breakage and repair for homologous recombination and the maintenance of oocyte integrity. Biallelic disruption of the DNA damage repair gene, <i>Fanconi anemia complementation group A</i> (<i>FANCA)</i>, is a common cause of Fanconi anaemia, a syndrome characterised by bone marrow failure, cancer predisposition, physical anomalies and POI. There is ongoing dispute about the role of heterozygous <i>FANCA</i> variants in POI pathogenesis, with insufficient evidence supporting causation. Here, we have identified biallelic <i>FANCA</i> variants in French sisters presenting with POI, including a novel missense variant of uncertain significance and a likely pathogenic deletion that initially evaded detection. Functional studies indicated no discernible effect on DNA damage sensitivity in patient lymphoblasts. These novel <i>FANCA</i> variants add evidence that heterozygous loss of one allele is insufficient to cause DNA damage sensitivity and POI. We propose that intragenic deletions, that are relatively common in <i>FANCA</i>, may be missed without careful analysis, and could explain the presumed causation of heterozygous variants. Accurate variant curation is critical to optimise patient care and outcomes.</p>\",\"PeriodicalId\":10354,\"journal\":{\"name\":\"Clinical Genetics\",\"volume\":\"106 3\",\"pages\":\"321-335\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2024-05-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14543\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cge.14543\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14543","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
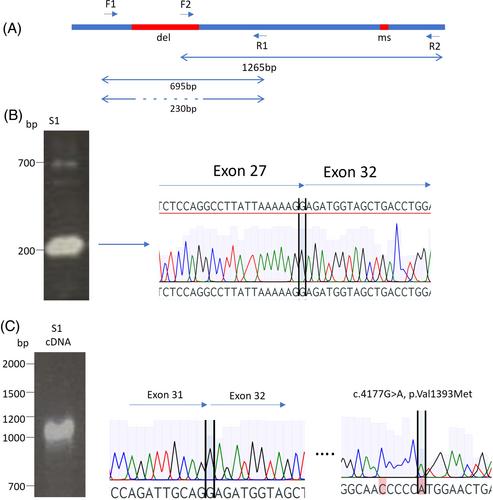
卵巢早衰是一种常见的女性不孕症,多达 4% 的女性会因此而不孕,其特点是 40 岁前闭经并伴有促性腺激素升高。卵母细胞需要受控的 DNA 断裂和修复,以进行同源重组和维持卵母细胞的完整性。DNA损伤修复基因--范可尼贫血补体A组(FANCA)的双偶缺失是范可尼贫血症的常见病因,这种综合征的特征是骨髓衰竭、癌症易感性、身体异常和POI。关于 FANCA 杂合子变异在 POI 发病机制中的作用,目前仍存在争议,没有足够的证据支持其因果关系。在这里,我们在出现 POI 的法国姐妹中发现了 FANCA 双重变异,包括一个意义不明的新型错义变异和一个最初未被发现的可能致病的缺失变异。功能研究表明,这些变异对患者淋巴母细胞的DNA损伤敏感性没有明显影响。这些新的 FANCA 变异补充了一个证据,即一个等位基因的杂合性缺失不足以导致 DNA 损伤敏感性和 POI。我们认为,基因内缺失在 FANCA 中比较常见,如果不仔细分析可能会漏掉,这也可以解释杂合变异的假定成因。准确的变异整理对于优化患者护理和预后至关重要。
Biallelic FANCA variants detected in sisters with isolated premature ovarian insufficiency
Premature ovarian insufficiency is a common form of female infertility affecting up to 4% of women and characterised by amenorrhea with elevated gonadotropin before the age of 40. Oocytes require controlled DNA breakage and repair for homologous recombination and the maintenance of oocyte integrity. Biallelic disruption of the DNA damage repair gene, Fanconi anemia complementation group A (FANCA), is a common cause of Fanconi anaemia, a syndrome characterised by bone marrow failure, cancer predisposition, physical anomalies and POI. There is ongoing dispute about the role of heterozygous FANCA variants in POI pathogenesis, with insufficient evidence supporting causation. Here, we have identified biallelic FANCA variants in French sisters presenting with POI, including a novel missense variant of uncertain significance and a likely pathogenic deletion that initially evaded detection. Functional studies indicated no discernible effect on DNA damage sensitivity in patient lymphoblasts. These novel FANCA variants add evidence that heterozygous loss of one allele is insufficient to cause DNA damage sensitivity and POI. We propose that intragenic deletions, that are relatively common in FANCA, may be missed without careful analysis, and could explain the presumed causation of heterozygous variants. Accurate variant curation is critical to optimise patient care and outcomes.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
