{"title":"将药物库化合物重新用作潜在的恶性疟原虫 1a 氨基酰 tRNA 合成酶多级泛抑制剂,特别关注丝裂霉素","authors":"Fisayo Olotu , Mariscal Brice Tchatat Tali , Curtis Chepsiror , Olivier Sheik Amamuddy , Fabrice Fekam Boyom , Özlem Tastan Bishop","doi":"10.1016/j.ijpddr.2024.100548","DOIUrl":null,"url":null,"abstract":"<div><p><em>Plasmodium falciparum</em> aminoacyl tRNA synthetases (PfaaRSs) are potent antimalarial targets essential for proteome fidelity and overall parasite survival in every stage of the parasite's life cycle. So far, some of these proteins have been singly targeted yielding inhibitor compounds that have been limited by incidences of resistance which can be overcome via pan-inhibition strategies. Hence, herein, for the first time, we report the identification and <em>in vitro</em> antiplasmodial validation of <strong>Mitomycin</strong> (<strong>MMC</strong>) as a probable pan-inhibitor of class 1a (arginyl(A)-, cysteinyl(C), isoleucyl(I)-, leucyl(L), methionyl(M), and valyl(V)-) PfaaRSs which hypothetically may underlie its previously reported activity on the ribosomal RNA to inhibit protein translation and biosynthesis. We combined multiple <em>in silico</em> structure-based discovery strategies that first helped identify functional and druggable sites that were preferentially targeted by the compound in each of the plasmodial proteins: Ins1-Ins2 domain in Pf-ARS; anticodon binding domain in Pf-CRS; CP1-editing domain in Pf-IRS and Pf-MRS; C-terminal domain in Pf-LRS; and CP-core region in Pf-VRS. Molecular dynamics studies further revealed that <strong>MMC</strong> allosterically induced changes in the global structures of each protein. Likewise, prominent structural perturbations were caused by the compound across the functional domains of the proteins. More so, <strong>MMC</strong> induced systematic alterations in the binding of the catalytic nucleotide and amino acid substrates which culminated in the loss of key interactions with key active site residues and ultimate reduction in the nucleotide-binding affinities across all proteins, as deduced from the binding energy calculations. These altogether confirmed that <strong>MMC</strong> uniformly disrupted the structure of the target proteins and essential substrates. Further, <strong>MMC</strong> demonstrated <em>IC</em><sub><em>50</em></sub> < 5 μM against the Dd2 and 3D7 strains of parasite making it a good starting point for malarial drug development. We believe that findings from our study will be important in the current search for highly effective multi-stage antimalarial drugs.</p></div>","PeriodicalId":13775,"journal":{"name":"International Journal for Parasitology: Drugs and Drug Resistance","volume":"25 ","pages":"Article 100548"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2211320724000290/pdfft?md5=6251aac0cd410b41f7cb2265f47c4b80&pid=1-s2.0-S2211320724000290-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Repurposing DrugBank compounds as potential Plasmodium falciparum class 1a aminoacyl tRNA synthetase multi-stage pan-inhibitors with a specific focus on mitomycin\",\"authors\":\"Fisayo Olotu , Mariscal Brice Tchatat Tali , Curtis Chepsiror , Olivier Sheik Amamuddy , Fabrice Fekam Boyom , Özlem Tastan Bishop\",\"doi\":\"10.1016/j.ijpddr.2024.100548\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p><em>Plasmodium falciparum</em> aminoacyl tRNA synthetases (PfaaRSs) are potent antimalarial targets essential for proteome fidelity and overall parasite survival in every stage of the parasite's life cycle. So far, some of these proteins have been singly targeted yielding inhibitor compounds that have been limited by incidences of resistance which can be overcome via pan-inhibition strategies. Hence, herein, for the first time, we report the identification and <em>in vitro</em> antiplasmodial validation of <strong>Mitomycin</strong> (<strong>MMC</strong>) as a probable pan-inhibitor of class 1a (arginyl(A)-, cysteinyl(C), isoleucyl(I)-, leucyl(L), methionyl(M), and valyl(V)-) PfaaRSs which hypothetically may underlie its previously reported activity on the ribosomal RNA to inhibit protein translation and biosynthesis. We combined multiple <em>in silico</em> structure-based discovery strategies that first helped identify functional and druggable sites that were preferentially targeted by the compound in each of the plasmodial proteins: Ins1-Ins2 domain in Pf-ARS; anticodon binding domain in Pf-CRS; CP1-editing domain in Pf-IRS and Pf-MRS; C-terminal domain in Pf-LRS; and CP-core region in Pf-VRS. Molecular dynamics studies further revealed that <strong>MMC</strong> allosterically induced changes in the global structures of each protein. Likewise, prominent structural perturbations were caused by the compound across the functional domains of the proteins. More so, <strong>MMC</strong> induced systematic alterations in the binding of the catalytic nucleotide and amino acid substrates which culminated in the loss of key interactions with key active site residues and ultimate reduction in the nucleotide-binding affinities across all proteins, as deduced from the binding energy calculations. These altogether confirmed that <strong>MMC</strong> uniformly disrupted the structure of the target proteins and essential substrates. Further, <strong>MMC</strong> demonstrated <em>IC</em><sub><em>50</em></sub> < 5 μM against the Dd2 and 3D7 strains of parasite making it a good starting point for malarial drug development. We believe that findings from our study will be important in the current search for highly effective multi-stage antimalarial drugs.</p></div>\",\"PeriodicalId\":13775,\"journal\":{\"name\":\"International Journal for Parasitology: Drugs and Drug Resistance\",\"volume\":\"25 \",\"pages\":\"Article 100548\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2211320724000290/pdfft?md5=6251aac0cd410b41f7cb2265f47c4b80&pid=1-s2.0-S2211320724000290-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal for Parasitology: Drugs and Drug Resistance\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2211320724000290\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/5/20 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"PARASITOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal for Parasitology: Drugs and Drug Resistance","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2211320724000290","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/20 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PARASITOLOGY","Score":null,"Total":0}
Repurposing DrugBank compounds as potential Plasmodium falciparum class 1a aminoacyl tRNA synthetase multi-stage pan-inhibitors with a specific focus on mitomycin
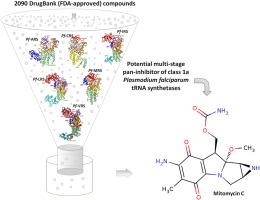
Plasmodium falciparum aminoacyl tRNA synthetases (PfaaRSs) are potent antimalarial targets essential for proteome fidelity and overall parasite survival in every stage of the parasite's life cycle. So far, some of these proteins have been singly targeted yielding inhibitor compounds that have been limited by incidences of resistance which can be overcome via pan-inhibition strategies. Hence, herein, for the first time, we report the identification and in vitro antiplasmodial validation of Mitomycin (MMC) as a probable pan-inhibitor of class 1a (arginyl(A)-, cysteinyl(C), isoleucyl(I)-, leucyl(L), methionyl(M), and valyl(V)-) PfaaRSs which hypothetically may underlie its previously reported activity on the ribosomal RNA to inhibit protein translation and biosynthesis. We combined multiple in silico structure-based discovery strategies that first helped identify functional and druggable sites that were preferentially targeted by the compound in each of the plasmodial proteins: Ins1-Ins2 domain in Pf-ARS; anticodon binding domain in Pf-CRS; CP1-editing domain in Pf-IRS and Pf-MRS; C-terminal domain in Pf-LRS; and CP-core region in Pf-VRS. Molecular dynamics studies further revealed that MMC allosterically induced changes in the global structures of each protein. Likewise, prominent structural perturbations were caused by the compound across the functional domains of the proteins. More so, MMC induced systematic alterations in the binding of the catalytic nucleotide and amino acid substrates which culminated in the loss of key interactions with key active site residues and ultimate reduction in the nucleotide-binding affinities across all proteins, as deduced from the binding energy calculations. These altogether confirmed that MMC uniformly disrupted the structure of the target proteins and essential substrates. Further, MMC demonstrated IC50 < 5 μM against the Dd2 and 3D7 strains of parasite making it a good starting point for malarial drug development. We believe that findings from our study will be important in the current search for highly effective multi-stage antimalarial drugs.
期刊介绍:
The International Journal for Parasitology – Drugs and Drug Resistance is one of a series of specialist, open access journals launched by the International Journal for Parasitology. It publishes the results of original research in the area of anti-parasite drug identification, development and evaluation, and parasite drug resistance. The journal also covers research into natural products as anti-parasitic agents, and bioactive parasite products. Studies can be aimed at unicellular or multicellular parasites of human or veterinary importance.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
