Friedrich Hastedt, Rowan M. Bailey, Klaus Hellgardt, Sophia N. Yaliraki, Ehecatl Antonio del Rio Chanona and Dongda Zhang
{"title":"研究用于化学逆合成的机器学习框架的可靠性和可解释性","authors":"Friedrich Hastedt, Rowan M. Bailey, Klaus Hellgardt, Sophia N. Yaliraki, Ehecatl Antonio del Rio Chanona and Dongda Zhang","doi":"10.1039/D4DD00007B","DOIUrl":null,"url":null,"abstract":"<p >Machine learning models for chemical retrosynthesis have attracted substantial interest in recent years. Unaddressed challenges, particularly the absence of robust evaluation metrics for performance comparison, and the lack of black-box interpretability, obscure model limitations and impede progress in the field. We present an automated benchmarking pipeline designed for effective model performance comparisons. With an emphasis on user-friendly design, we aim to streamline accessibility and facilitate utilisation within the research community. Additionally, we suggest and perform a new interpretability study to uncover the degree of chemical understanding acquired by retrosynthesis models. Our results reveal that frameworks based on chemical reaction rules yield the most diverse, chemically valid, and feasible reactions, whereas purely data-driven frameworks suffer from unfeasible and invalid predictions. The interpretability study emphasises that incorporating reaction rules not only enhances model performance but also improves interpretability. For simple molecules, we show that Graph Neural Networks identify relevant functional groups in the product molecule, offering model interpretability. Sequence-to-sequence Transformers are not found to provide such an explanation. As the molecule and reaction mechanism grow more complex, both data-driven models propose unfeasible disconnections without offering a chemical rationale. We stress the importance of incorporating chemically meaningful descriptors within deep-learning models. Our study provides valuable guidance for the future development of retrosynthesis frameworks.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":null,"pages":null},"PeriodicalIF":6.2000,"publicationDate":"2024-05-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d4dd00007b?page=search","citationCount":"0","resultStr":"{\"title\":\"Investigating the reliability and interpretability of machine learning frameworks for chemical retrosynthesis†\",\"authors\":\"Friedrich Hastedt, Rowan M. Bailey, Klaus Hellgardt, Sophia N. Yaliraki, Ehecatl Antonio del Rio Chanona and Dongda Zhang\",\"doi\":\"10.1039/D4DD00007B\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Machine learning models for chemical retrosynthesis have attracted substantial interest in recent years. Unaddressed challenges, particularly the absence of robust evaluation metrics for performance comparison, and the lack of black-box interpretability, obscure model limitations and impede progress in the field. We present an automated benchmarking pipeline designed for effective model performance comparisons. With an emphasis on user-friendly design, we aim to streamline accessibility and facilitate utilisation within the research community. Additionally, we suggest and perform a new interpretability study to uncover the degree of chemical understanding acquired by retrosynthesis models. Our results reveal that frameworks based on chemical reaction rules yield the most diverse, chemically valid, and feasible reactions, whereas purely data-driven frameworks suffer from unfeasible and invalid predictions. The interpretability study emphasises that incorporating reaction rules not only enhances model performance but also improves interpretability. For simple molecules, we show that Graph Neural Networks identify relevant functional groups in the product molecule, offering model interpretability. Sequence-to-sequence Transformers are not found to provide such an explanation. As the molecule and reaction mechanism grow more complex, both data-driven models propose unfeasible disconnections without offering a chemical rationale. We stress the importance of incorporating chemically meaningful descriptors within deep-learning models. Our study provides valuable guidance for the future development of retrosynthesis frameworks.</p>\",\"PeriodicalId\":72816,\"journal\":{\"name\":\"Digital discovery\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":6.2000,\"publicationDate\":\"2024-05-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d4dd00007b?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Digital discovery\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d4dd00007b\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d4dd00007b","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
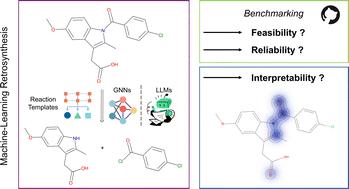
Investigating the reliability and interpretability of machine learning frameworks for chemical retrosynthesis†
Machine learning models for chemical retrosynthesis have attracted substantial interest in recent years. Unaddressed challenges, particularly the absence of robust evaluation metrics for performance comparison, and the lack of black-box interpretability, obscure model limitations and impede progress in the field. We present an automated benchmarking pipeline designed for effective model performance comparisons. With an emphasis on user-friendly design, we aim to streamline accessibility and facilitate utilisation within the research community. Additionally, we suggest and perform a new interpretability study to uncover the degree of chemical understanding acquired by retrosynthesis models. Our results reveal that frameworks based on chemical reaction rules yield the most diverse, chemically valid, and feasible reactions, whereas purely data-driven frameworks suffer from unfeasible and invalid predictions. The interpretability study emphasises that incorporating reaction rules not only enhances model performance but also improves interpretability. For simple molecules, we show that Graph Neural Networks identify relevant functional groups in the product molecule, offering model interpretability. Sequence-to-sequence Transformers are not found to provide such an explanation. As the molecule and reaction mechanism grow more complex, both data-driven models propose unfeasible disconnections without offering a chemical rationale. We stress the importance of incorporating chemically meaningful descriptors within deep-learning models. Our study provides valuable guidance for the future development of retrosynthesis frameworks.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
