{"title":"开发基于基本反应的动力学模型,以预测反应性自由基诱导的有机化合物的水相命运。","authors":"Daisuke Minakata*, ","doi":"10.1021/acs.accounts.4c00021","DOIUrl":null,"url":null,"abstract":"<p >Aqueous-phase free radicals such as reactive oxygen, halogen, and nitrogen species play important roles in the fate of organic compounds in the aqueous-phase advanced water treatment processes and natural aquatic environments under sunlight irradiation. Predicting the fate of organic compounds in aqueous-phase advanced water treatment processes and natural aquatic environments necessitates understanding the kinetics and reaction mechanisms of initial reactions of free radicals with structurally diverse organic compounds and other reactions. Researchers developed conventional predictive models based on experimentally measured transformation products and determined reaction rate constants by fitting with the time-dependent concentration profiles of species due to difficulties in their measurements of unstable intermediates. However, the empirical treatment of lumped reaction mechanisms had a model prediction limitation with respect to the specific parent compound’s fate. We use <i>ab initio</i> and density functional theory quantum chemical computations, numerical solutions of ordinary differential equations, and validation of the outcomes of the model with experiments. Sensitivity analysis of reaction rate constants and concentration profiles enables us to identify an important elementary reaction in formating the transformation product. Such predictive elementary reaction-based kinetics models can be used to screen organic compounds in water and predict their potentially toxic transformation products for a specific experimental investigation.</p><p >Over the past decade, we determined linear free energy relationships (LFERs) that bridge the kinetic and thermochemical properties of reactive oxygen species such as hydroxyl radicals (HO<sup>•</sup>), peroxyl radicals (ROO<sup>•</sup>), and singlet oxygen (<sup>1</sup>O<sub>2</sub>); reactive halogen species such as chlorine radicals (Cl<sup>•</sup>) and bromine radicals (Br<sup>•</sup>); reactive nitrogen species (NO<sub>2</sub><sup>•</sup>); and carbonate radicals (CO<sub>3</sub><sup>•–</sup>). We used literature-reported experimental rate constants as kinetic information. We considered the theoretically calculated aqueous-phase free energy of activation or reaction to be a kinetic or a thermochemical property, and obtained via validated <i>ab initio</i> or density functional theory-based quantum chemical computations using explicit and implicit solvation models. We determined rate-determining reaction mechanisms involved in reactions by observing robust LFERs. The general accuracy of LFERs to predict aqueous-phase rate constants was within a difference of a factor of 2–5 from experimental values.</p><p >We developed elementary reaction-based kinetic models and predicted the fate of acetone induced by HO<sup>•</sup> in an advanced water treatment process and methionine by photochemically produced reactive intermediates in sunlit fresh waters. We provided mechanistic insight into peroxyl radical reaction mechanisms and critical roles in the degradation of acetone and the formation of transformation products. We highlighted different roles of triplet excited states of two surrogate CDOMs, <sup>1</sup>O<sub>2</sub>, and HO<sup>•</sup>, in methionine degradation. Predicted transformation products were compared to those obtained via benchtop experiments to validate our elementary reaction-based kinetic models. Predicting the reactivities of reactive halogen and nitrogen species implicates our understanding of the formation of potentially toxic halogen- and nitrogen-containing transformation products during water treatment processes and in natural aquatic environments.</p>","PeriodicalId":1,"journal":{"name":"Accounts of Chemical Research","volume":"57 12","pages":"1658–1669"},"PeriodicalIF":17.7000,"publicationDate":"2024-05-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Development of an Elementary Reaction-Based Kinetic Model to Predict the Aqueous-Phase Fate of Organic Compounds Induced by Reactive Free Radicals\",\"authors\":\"Daisuke Minakata*, \",\"doi\":\"10.1021/acs.accounts.4c00021\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Aqueous-phase free radicals such as reactive oxygen, halogen, and nitrogen species play important roles in the fate of organic compounds in the aqueous-phase advanced water treatment processes and natural aquatic environments under sunlight irradiation. Predicting the fate of organic compounds in aqueous-phase advanced water treatment processes and natural aquatic environments necessitates understanding the kinetics and reaction mechanisms of initial reactions of free radicals with structurally diverse organic compounds and other reactions. Researchers developed conventional predictive models based on experimentally measured transformation products and determined reaction rate constants by fitting with the time-dependent concentration profiles of species due to difficulties in their measurements of unstable intermediates. However, the empirical treatment of lumped reaction mechanisms had a model prediction limitation with respect to the specific parent compound’s fate. We use <i>ab initio</i> and density functional theory quantum chemical computations, numerical solutions of ordinary differential equations, and validation of the outcomes of the model with experiments. Sensitivity analysis of reaction rate constants and concentration profiles enables us to identify an important elementary reaction in formating the transformation product. Such predictive elementary reaction-based kinetics models can be used to screen organic compounds in water and predict their potentially toxic transformation products for a specific experimental investigation.</p><p >Over the past decade, we determined linear free energy relationships (LFERs) that bridge the kinetic and thermochemical properties of reactive oxygen species such as hydroxyl radicals (HO<sup>•</sup>), peroxyl radicals (ROO<sup>•</sup>), and singlet oxygen (<sup>1</sup>O<sub>2</sub>); reactive halogen species such as chlorine radicals (Cl<sup>•</sup>) and bromine radicals (Br<sup>•</sup>); reactive nitrogen species (NO<sub>2</sub><sup>•</sup>); and carbonate radicals (CO<sub>3</sub><sup>•–</sup>). We used literature-reported experimental rate constants as kinetic information. We considered the theoretically calculated aqueous-phase free energy of activation or reaction to be a kinetic or a thermochemical property, and obtained via validated <i>ab initio</i> or density functional theory-based quantum chemical computations using explicit and implicit solvation models. We determined rate-determining reaction mechanisms involved in reactions by observing robust LFERs. The general accuracy of LFERs to predict aqueous-phase rate constants was within a difference of a factor of 2–5 from experimental values.</p><p >We developed elementary reaction-based kinetic models and predicted the fate of acetone induced by HO<sup>•</sup> in an advanced water treatment process and methionine by photochemically produced reactive intermediates in sunlit fresh waters. We provided mechanistic insight into peroxyl radical reaction mechanisms and critical roles in the degradation of acetone and the formation of transformation products. We highlighted different roles of triplet excited states of two surrogate CDOMs, <sup>1</sup>O<sub>2</sub>, and HO<sup>•</sup>, in methionine degradation. Predicted transformation products were compared to those obtained via benchtop experiments to validate our elementary reaction-based kinetic models. Predicting the reactivities of reactive halogen and nitrogen species implicates our understanding of the formation of potentially toxic halogen- and nitrogen-containing transformation products during water treatment processes and in natural aquatic environments.</p>\",\"PeriodicalId\":1,\"journal\":{\"name\":\"Accounts of Chemical Research\",\"volume\":\"57 12\",\"pages\":\"1658–1669\"},\"PeriodicalIF\":17.7000,\"publicationDate\":\"2024-05-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Accounts of Chemical Research\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.accounts.4c00021\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Accounts of Chemical Research","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.accounts.4c00021","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
内容摘要 水相自由基(如活性氧、卤素和氮物种)在阳光照射下对水相高级水处理工艺和天然水生环境中有机化合物的归宿起着重要作用。要预测有机化合物在水相先进水处理工艺和天然水生环境中的归宿,就必须了解自由基与结构各异的有机化合物发生初始反应和其他反应的动力学和反应机理。由于难以测量不稳定的中间产物,研究人员根据实验测量的转化产物建立了传统的预测模型,并通过拟合随时间变化的物种浓度曲线来确定反应速率常数。然而,对成组反应机理的经验处理在特定母体化合物的命运方面存在模型预测的局限性。我们采用了原子序数和密度泛函理论量子化学计算、常微分方程数值解法,并通过实验对模型结果进行了验证。通过对反应速率常数和浓度曲线的敏感性分析,我们确定了形成转化产物的重要基本反应。这种基于基本反应的预测性动力学模型可用于筛选水中的有机化合物,并预测其潜在的毒性转化产物,以进行特定的实验研究。在过去的十年中,我们确定了线性自由能关系(LFERs),它连接了活性氧物种(如羟自由基(HO-)、过氧自由基(ROO-)和单线态氧(1O2))、活性卤素物种(如氯自由基(Cl-)和溴自由基(Br-))、活性氮物种(NO2-)和碳酸自由基(CO3-)的动力学和热化学性质。我们使用文献报告的实验速率常数作为动力学信息。我们认为理论计算出的水相活化自由能或反应自由能是一种动力学或热化学性质,是通过有效的 ab initio 或基于密度泛函理论的量子化学计算,使用显式和隐式溶解模型获得的。我们通过观察稳健的 LFER 确定了反应中的速率决定反应机制。我们建立了基于基本反应的动力学模型,并预测了在先进水处理工艺中由 HO- 诱导的丙酮和在日照淡水中由光化学产生的反应中间体诱导的蛋氨酸的归宿。我们从机理上深入探讨了过氧自由基反应机制以及在丙酮降解和转化产物形成过程中的关键作用。我们强调了 1O2 和 HO- 这两种替代性 CDOM 的三重激发态在蛋氨酸降解过程中的不同作用。我们将预测的转化产物与通过台式实验获得的产物进行了比较,以验证我们基于基本反应的动力学模型。预测活性卤素和氮物种的反应活性有助于我们了解在水处理过程和自然水生环境中形成的具有潜在毒性的含卤素和含氮转化产物。
Development of an Elementary Reaction-Based Kinetic Model to Predict the Aqueous-Phase Fate of Organic Compounds Induced by Reactive Free Radicals
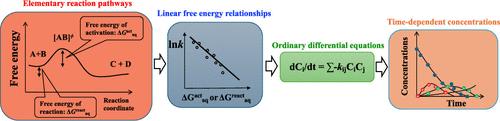
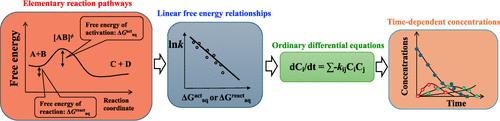
Aqueous-phase free radicals such as reactive oxygen, halogen, and nitrogen species play important roles in the fate of organic compounds in the aqueous-phase advanced water treatment processes and natural aquatic environments under sunlight irradiation. Predicting the fate of organic compounds in aqueous-phase advanced water treatment processes and natural aquatic environments necessitates understanding the kinetics and reaction mechanisms of initial reactions of free radicals with structurally diverse organic compounds and other reactions. Researchers developed conventional predictive models based on experimentally measured transformation products and determined reaction rate constants by fitting with the time-dependent concentration profiles of species due to difficulties in their measurements of unstable intermediates. However, the empirical treatment of lumped reaction mechanisms had a model prediction limitation with respect to the specific parent compound’s fate. We use ab initio and density functional theory quantum chemical computations, numerical solutions of ordinary differential equations, and validation of the outcomes of the model with experiments. Sensitivity analysis of reaction rate constants and concentration profiles enables us to identify an important elementary reaction in formating the transformation product. Such predictive elementary reaction-based kinetics models can be used to screen organic compounds in water and predict their potentially toxic transformation products for a specific experimental investigation.
Over the past decade, we determined linear free energy relationships (LFERs) that bridge the kinetic and thermochemical properties of reactive oxygen species such as hydroxyl radicals (HO•), peroxyl radicals (ROO•), and singlet oxygen (1O2); reactive halogen species such as chlorine radicals (Cl•) and bromine radicals (Br•); reactive nitrogen species (NO2•); and carbonate radicals (CO3•–). We used literature-reported experimental rate constants as kinetic information. We considered the theoretically calculated aqueous-phase free energy of activation or reaction to be a kinetic or a thermochemical property, and obtained via validated ab initio or density functional theory-based quantum chemical computations using explicit and implicit solvation models. We determined rate-determining reaction mechanisms involved in reactions by observing robust LFERs. The general accuracy of LFERs to predict aqueous-phase rate constants was within a difference of a factor of 2–5 from experimental values.
We developed elementary reaction-based kinetic models and predicted the fate of acetone induced by HO• in an advanced water treatment process and methionine by photochemically produced reactive intermediates in sunlit fresh waters. We provided mechanistic insight into peroxyl radical reaction mechanisms and critical roles in the degradation of acetone and the formation of transformation products. We highlighted different roles of triplet excited states of two surrogate CDOMs, 1O2, and HO•, in methionine degradation. Predicted transformation products were compared to those obtained via benchtop experiments to validate our elementary reaction-based kinetic models. Predicting the reactivities of reactive halogen and nitrogen species implicates our understanding of the formation of potentially toxic halogen- and nitrogen-containing transformation products during water treatment processes and in natural aquatic environments.
期刊介绍:
Accounts of Chemical Research presents short, concise and critical articles offering easy-to-read overviews of basic research and applications in all areas of chemistry and biochemistry. These short reviews focus on research from the author’s own laboratory and are designed to teach the reader about a research project. In addition, Accounts of Chemical Research publishes commentaries that give an informed opinion on a current research problem. Special Issues online are devoted to a single topic of unusual activity and significance.
Accounts of Chemical Research replaces the traditional article abstract with an article "Conspectus." These entries synopsize the research affording the reader a closer look at the content and significance of an article. Through this provision of a more detailed description of the article contents, the Conspectus enhances the article's discoverability by search engines and the exposure for the research.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
