Huairen Zhang, Avgi Andreou, Rupesh Bhatt, James Whitworth, Bryndis Yngvadottir, Eamonn R. Maher
{"title":"多发性原发性肾肿瘤的特征、病因及其对治疗的影响:系统综述。","authors":"Huairen Zhang, Avgi Andreou, Rupesh Bhatt, James Whitworth, Bryndis Yngvadottir, Eamonn R. Maher","doi":"10.1038/s41431-024-01628-5","DOIUrl":null,"url":null,"abstract":"In a subset of patients with renal tumours, multiple primary lesions may occur. Predisposition to multiple primary renal tumours (MPRT) is a well-recognised feature of some inherited renal cancer syndromes. The diagnosis of MPRT should therefore provoke a thorough assessment for clinical and genetic evidence of disorders associated with predisposition to renal tumourigenesis. To better define the clinical and genetic characteristics of MPRT, a systematic literature review was performed for publications up to 3 April 2024. A total of 7689 patients from 467 articles were identified with MPRT. Compared to all patients with renal cell carcinoma (RCC), patients with MPRT were more likely to be male (71.8% versus 63%) and have an earlier age at diagnosis (<46 years, 32.4% versus 19%). In 61.1% of cases MPRT were synchronous. The proportion of cases with similar histology and the proportion of cases with multiple papillary renal cell carcinoma (RCC) (16.1%) were higher than expected. In total, 14.9% of patients with MPRT had a family history of cancer or were diagnosed with a hereditary RCC associated syndrome with von Hippel-Lindau (VHL) disease being the most common one (69.7%), followed by Birt-Hogg-Dubé (BHD) syndrome (14.2%). Individuals with a known or likely genetic cause were, on average, younger (43.9 years versus 57.1 years). In rare cases intrarenal metastatic RCC can phenocopy MPRT. We review potential genetic causes of MPRT and their implications for management, suggest an approach to genetic testing for individuals presenting with MPRT and considerations in cases in which routine germline genetic testing does not provide a diagnosis.","PeriodicalId":12016,"journal":{"name":"European Journal of Human Genetics","volume":"32 8","pages":"887-894"},"PeriodicalIF":4.6000,"publicationDate":"2024-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11291654/pdf/","citationCount":"0","resultStr":"{\"title\":\"Characteristics, aetiology and implications for management of multiple primary renal tumours: a systematic review\",\"authors\":\"Huairen Zhang, Avgi Andreou, Rupesh Bhatt, James Whitworth, Bryndis Yngvadottir, Eamonn R. Maher\",\"doi\":\"10.1038/s41431-024-01628-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"In a subset of patients with renal tumours, multiple primary lesions may occur. Predisposition to multiple primary renal tumours (MPRT) is a well-recognised feature of some inherited renal cancer syndromes. The diagnosis of MPRT should therefore provoke a thorough assessment for clinical and genetic evidence of disorders associated with predisposition to renal tumourigenesis. To better define the clinical and genetic characteristics of MPRT, a systematic literature review was performed for publications up to 3 April 2024. A total of 7689 patients from 467 articles were identified with MPRT. Compared to all patients with renal cell carcinoma (RCC), patients with MPRT were more likely to be male (71.8% versus 63%) and have an earlier age at diagnosis (<46 years, 32.4% versus 19%). In 61.1% of cases MPRT were synchronous. The proportion of cases with similar histology and the proportion of cases with multiple papillary renal cell carcinoma (RCC) (16.1%) were higher than expected. In total, 14.9% of patients with MPRT had a family history of cancer or were diagnosed with a hereditary RCC associated syndrome with von Hippel-Lindau (VHL) disease being the most common one (69.7%), followed by Birt-Hogg-Dubé (BHD) syndrome (14.2%). Individuals with a known or likely genetic cause were, on average, younger (43.9 years versus 57.1 years). In rare cases intrarenal metastatic RCC can phenocopy MPRT. We review potential genetic causes of MPRT and their implications for management, suggest an approach to genetic testing for individuals presenting with MPRT and considerations in cases in which routine germline genetic testing does not provide a diagnosis.\",\"PeriodicalId\":12016,\"journal\":{\"name\":\"European Journal of Human Genetics\",\"volume\":\"32 8\",\"pages\":\"887-894\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2024-05-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11291654/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"European Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s41431-024-01628-5\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41431-024-01628-5","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Characteristics, aetiology and implications for management of multiple primary renal tumours: a systematic review
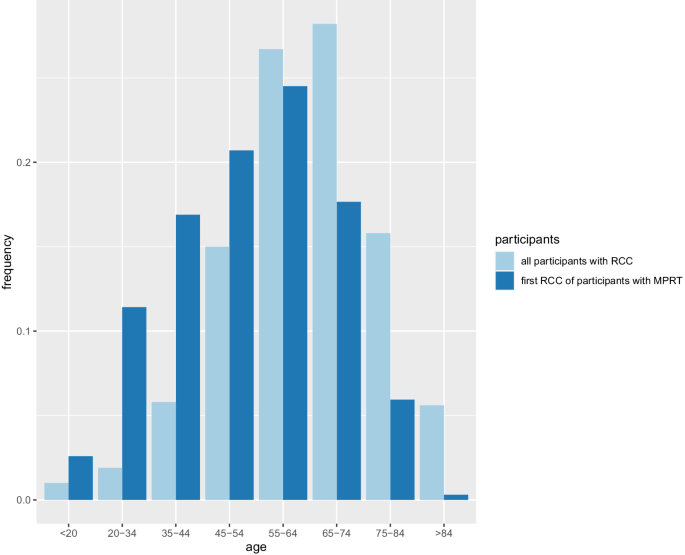
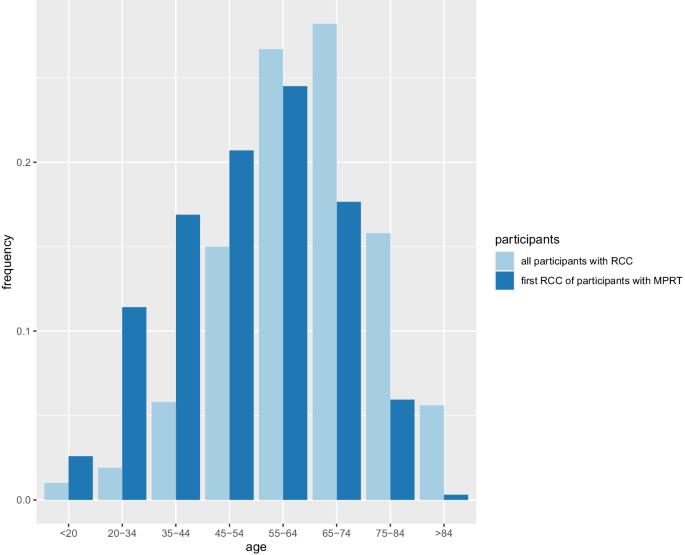
In a subset of patients with renal tumours, multiple primary lesions may occur. Predisposition to multiple primary renal tumours (MPRT) is a well-recognised feature of some inherited renal cancer syndromes. The diagnosis of MPRT should therefore provoke a thorough assessment for clinical and genetic evidence of disorders associated with predisposition to renal tumourigenesis. To better define the clinical and genetic characteristics of MPRT, a systematic literature review was performed for publications up to 3 April 2024. A total of 7689 patients from 467 articles were identified with MPRT. Compared to all patients with renal cell carcinoma (RCC), patients with MPRT were more likely to be male (71.8% versus 63%) and have an earlier age at diagnosis (<46 years, 32.4% versus 19%). In 61.1% of cases MPRT were synchronous. The proportion of cases with similar histology and the proportion of cases with multiple papillary renal cell carcinoma (RCC) (16.1%) were higher than expected. In total, 14.9% of patients with MPRT had a family history of cancer or were diagnosed with a hereditary RCC associated syndrome with von Hippel-Lindau (VHL) disease being the most common one (69.7%), followed by Birt-Hogg-Dubé (BHD) syndrome (14.2%). Individuals with a known or likely genetic cause were, on average, younger (43.9 years versus 57.1 years). In rare cases intrarenal metastatic RCC can phenocopy MPRT. We review potential genetic causes of MPRT and their implications for management, suggest an approach to genetic testing for individuals presenting with MPRT and considerations in cases in which routine germline genetic testing does not provide a diagnosis.
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
