{"title":"利用无监督机器学习降低维度,对二氧化碳-水界面进行 Ab Initio 表征。","authors":"Tetsuya Morishita*, and , Masashige Shiga, ","doi":"10.1021/acs.jpcb.4c01526","DOIUrl":null,"url":null,"abstract":"<p >Precise characterization of the supercritical CO<sub>2</sub>–water interface under high pressure and temperature conditions is crucial for the geological storage of carbon dioxide (CO<sub>2</sub>) in deep saline aquifers. Molecular dynamics (MD) simulations offer a valuable approach to gaining insight into the CO<sub>2</sub>–water interface at a microscopic level. However, no attempt has been made to characterize the CO<sub>2</sub>–water interface with the accuracy afforded by <i>ab initio</i> calculations. In this study, we performed <i>ab initio</i> MD (AIMD) simulations to investigate the structural and dynamical properties of the CO<sub>2</sub>–water interface, comparing the results with those obtained from classical force-field MD (FF-MD) simulations. Molecular orientation at the interface was well reproduced in both AIMD and FF-MD simulations. Characteristic structural fluctuations of water at the interface were unveiled by applying multidimensional scaling and time-dependent principal component analysis to the AIMD trajectories; however, they were not prominent in the FF-MD simulations. Furthermore, our study demonstrated a marked difference in the residence time of molecules in the interface region between AIMD and FF-MD simulations, indicating that time-dependent properties of the CO<sub>2</sub>–water interface strongly depend on the description of the intermolecular forces.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":"128 23","pages":"5781–5791"},"PeriodicalIF":2.9000,"publicationDate":"2024-06-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Ab Initio Characterization of the CO2–Water Interface Using Unsupervised Machine Learning for Dimensionality Reduction\",\"authors\":\"Tetsuya Morishita*, and , Masashige Shiga, \",\"doi\":\"10.1021/acs.jpcb.4c01526\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Precise characterization of the supercritical CO<sub>2</sub>–water interface under high pressure and temperature conditions is crucial for the geological storage of carbon dioxide (CO<sub>2</sub>) in deep saline aquifers. Molecular dynamics (MD) simulations offer a valuable approach to gaining insight into the CO<sub>2</sub>–water interface at a microscopic level. However, no attempt has been made to characterize the CO<sub>2</sub>–water interface with the accuracy afforded by <i>ab initio</i> calculations. In this study, we performed <i>ab initio</i> MD (AIMD) simulations to investigate the structural and dynamical properties of the CO<sub>2</sub>–water interface, comparing the results with those obtained from classical force-field MD (FF-MD) simulations. Molecular orientation at the interface was well reproduced in both AIMD and FF-MD simulations. Characteristic structural fluctuations of water at the interface were unveiled by applying multidimensional scaling and time-dependent principal component analysis to the AIMD trajectories; however, they were not prominent in the FF-MD simulations. Furthermore, our study demonstrated a marked difference in the residence time of molecules in the interface region between AIMD and FF-MD simulations, indicating that time-dependent properties of the CO<sub>2</sub>–water interface strongly depend on the description of the intermolecular forces.</p>\",\"PeriodicalId\":60,\"journal\":{\"name\":\"The Journal of Physical Chemistry B\",\"volume\":\"128 23\",\"pages\":\"5781–5791\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-06-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry B\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcb.4c01526\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcb.4c01526","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
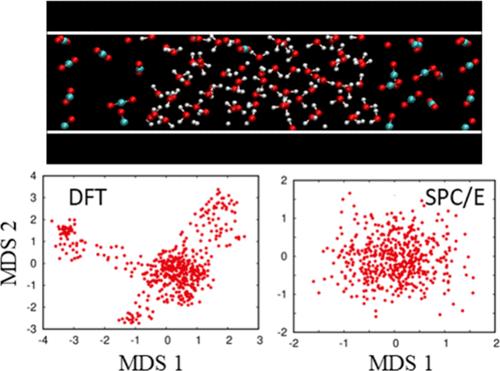
高压和高温条件下超临界二氧化碳-水界面的精确表征对于二氧化碳(CO2)在深盐含水层中的地质封存至关重要。分子动力学(MD)模拟为从微观层面深入了解二氧化碳-水界面提供了宝贵的方法。然而,目前还没有人尝试过用原子序数计算所提供的精度来描述二氧化碳-水界面。在本研究中,我们进行了 ab initio MD(AIMD)模拟,以研究二氧化碳-水界面的结构和动力学特性,并将结果与经典力场 MD(FF-MD)模拟的结果进行了比较。AIMD 和 FF-MD 模拟都很好地再现了界面上的分子取向。通过对 AIMD 轨迹进行多维缩放和随时间变化的主成分分析,揭示了界面处水的特征性结构波动;然而,这些波动在 FF-MD 模拟中并不突出。此外,我们的研究表明,在 AIMD 和 FF-MD 模拟中,分子在界面区域的停留时间存在明显差异,这表明二氧化碳-水界面随时间变化的特性在很大程度上取决于对分子间作用力的描述。
Ab Initio Characterization of the CO2–Water Interface Using Unsupervised Machine Learning for Dimensionality Reduction
Precise characterization of the supercritical CO2–water interface under high pressure and temperature conditions is crucial for the geological storage of carbon dioxide (CO2) in deep saline aquifers. Molecular dynamics (MD) simulations offer a valuable approach to gaining insight into the CO2–water interface at a microscopic level. However, no attempt has been made to characterize the CO2–water interface with the accuracy afforded by ab initio calculations. In this study, we performed ab initio MD (AIMD) simulations to investigate the structural and dynamical properties of the CO2–water interface, comparing the results with those obtained from classical force-field MD (FF-MD) simulations. Molecular orientation at the interface was well reproduced in both AIMD and FF-MD simulations. Characteristic structural fluctuations of water at the interface were unveiled by applying multidimensional scaling and time-dependent principal component analysis to the AIMD trajectories; however, they were not prominent in the FF-MD simulations. Furthermore, our study demonstrated a marked difference in the residence time of molecules in the interface region between AIMD and FF-MD simulations, indicating that time-dependent properties of the CO2–water interface strongly depend on the description of the intermolecular forces.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
