D. Christopher Braddock, Siwoo Lee and Henry S. Rzepa
{"title":"模拟 SWERN 氧化的动力学同位素效应。基于 DFT 的过渡态理论没问题。","authors":"D. Christopher Braddock, Siwoo Lee and Henry S. Rzepa","doi":"10.1039/D3DD00246B","DOIUrl":null,"url":null,"abstract":"<p >We investigate the model reported by Giagou and Meyer in 2010 for comparing deuterium kinetic isotope effects (KIEs) computed using density functional theory (DFT) for the intramolecular hydrogen transfer step in the mechanism of the Swern oxidation of alcohols to aldehydes, with those measured by experiment. Whereas the replication of the original computed values for the gas-phase reaction proved entirely successful, several issues were discovered when a continuum solvent model was used. These included uncertainty regarding the parameters and methods used for the calculations and also the coordinates for the original reactant and transition structures, <em>via</em> their provision as data in the ESI. The original conclusions, in which a numerical mis-match between the magnitude of the computed and experimentally measured KIE was attributed to significant deviations from transition structure theory, are here instead rationalised as a manifestation of basis-set effects in the computation. Transition state theory appears to be operating successfully. We now recommend the use of basis sets of triple- or quadruple-ζ quality, rather than the split-valence level previously employed, that dispersion energy corrections be included and that a continuum solvent model using smoothed reaction cavities is essential for effective geometry optimisation and hence accurate normal coordinate analysis. An outlying experimental KIE obtained for chloroform as solvent is attributed to a small level of an explicit hydrogen bonded interaction with the substrate. A temperature outlier for the measured KIE at 195 K is suggested for further experimental investigation, although it may also be an indication of an unusually abrupt incursion of hydrogen tunnelling which would need non-Born–Oppenheimer methods in which nuclear quantum effects are included to be more accurately modelled. We predict KIEs for new substituents, of which those for R = NMe<small><sub>2</sub></small> are significantly larger than those for R = H. This approach could be useful in designing variations of the Swern reagent that could lead to synthesis of aldehydes incorporating much higher levels of deuterium. The use of FAIR data rather than the traditional model of its inclusion in the ESI is discussed, and two data discovery tools exploiting these FAIR attributes are suggested.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 8","pages":" 1496-1508"},"PeriodicalIF":6.2000,"publicationDate":"2024-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d3dd00246b?page=search","citationCount":"0","resultStr":"{\"title\":\"Modelling kinetic isotope effects for Swern oxidation using DFT-based transition state theory†\",\"authors\":\"D. Christopher Braddock, Siwoo Lee and Henry S. Rzepa\",\"doi\":\"10.1039/D3DD00246B\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We investigate the model reported by Giagou and Meyer in 2010 for comparing deuterium kinetic isotope effects (KIEs) computed using density functional theory (DFT) for the intramolecular hydrogen transfer step in the mechanism of the Swern oxidation of alcohols to aldehydes, with those measured by experiment. Whereas the replication of the original computed values for the gas-phase reaction proved entirely successful, several issues were discovered when a continuum solvent model was used. These included uncertainty regarding the parameters and methods used for the calculations and also the coordinates for the original reactant and transition structures, <em>via</em> their provision as data in the ESI. The original conclusions, in which a numerical mis-match between the magnitude of the computed and experimentally measured KIE was attributed to significant deviations from transition structure theory, are here instead rationalised as a manifestation of basis-set effects in the computation. Transition state theory appears to be operating successfully. We now recommend the use of basis sets of triple- or quadruple-ζ quality, rather than the split-valence level previously employed, that dispersion energy corrections be included and that a continuum solvent model using smoothed reaction cavities is essential for effective geometry optimisation and hence accurate normal coordinate analysis. An outlying experimental KIE obtained for chloroform as solvent is attributed to a small level of an explicit hydrogen bonded interaction with the substrate. A temperature outlier for the measured KIE at 195 K is suggested for further experimental investigation, although it may also be an indication of an unusually abrupt incursion of hydrogen tunnelling which would need non-Born–Oppenheimer methods in which nuclear quantum effects are included to be more accurately modelled. We predict KIEs for new substituents, of which those for R = NMe<small><sub>2</sub></small> are significantly larger than those for R = H. This approach could be useful in designing variations of the Swern reagent that could lead to synthesis of aldehydes incorporating much higher levels of deuterium. The use of FAIR data rather than the traditional model of its inclusion in the ESI is discussed, and two data discovery tools exploiting these FAIR attributes are suggested.</p>\",\"PeriodicalId\":72816,\"journal\":{\"name\":\"Digital discovery\",\"volume\":\" 8\",\"pages\":\" 1496-1508\"},\"PeriodicalIF\":6.2000,\"publicationDate\":\"2024-06-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d3dd00246b?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Digital discovery\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d3dd00246b\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d3dd00246b","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
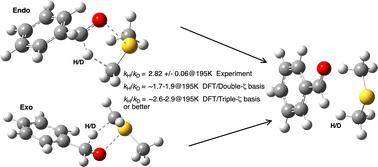
我们对 Giagou 和 Meyer 于 2010 年报告的模型进行了研究,以比较使用 DFT 理论计算的醇氧化成醛机理中分子内氢转移步骤的氘动力学同位素效应(KIE)与实验测量值。事实证明,复制气相反应的原始计算值是完全成功的,但在使用连续溶剂模型时发现了一些问题。这些问题包括计算所用参数和方法的不确定性,以及通过在电子辅助信息(ESI)中作为数据提供的原始反应物和过渡态的坐标。原来的结论认为,计算得出的 KIE 与实验测量得出的 KIE 在数值上的不匹配是由于严重偏离了过渡态理论,而这里的结论则被合理地解释为计算中基集效应的表现。过渡态理论似乎运行成功。我们现在建议使用三重或四重ζ质量的基集,而不是之前使用的分价水平,并建议将色散能校正包括在内,而且使用平滑反应腔的连续溶剂模型对于有效的几何优化以及精确的法线坐标分析至关重要。以氯仿为溶剂的实验 KIE 值偏离值较小,这是因为与基质之间存在少量明确的氢键相互作用。我们建议对 195K 时测得的 KIE 的温度离群值进行进一步的实验研究,不过这也可能是氢隧穿异常突然侵入的一种迹象,这就需要采用包含核量子效应的非 Born-Oppenheimer 方法来进行更精确的建模。我们预测了新取代基的 KIE,其中 R=NMe2 的 KIE 明显大于 R=H 的 KIE。这种方法有助于设计 Swern 试剂的变体,从而合成氘含量更高的醛。本文讨论了 FAIR 数据的使用,而不是将其纳入电子辅助信息 (ESI) 的传统模式。
Modelling kinetic isotope effects for Swern oxidation using DFT-based transition state theory†
We investigate the model reported by Giagou and Meyer in 2010 for comparing deuterium kinetic isotope effects (KIEs) computed using density functional theory (DFT) for the intramolecular hydrogen transfer step in the mechanism of the Swern oxidation of alcohols to aldehydes, with those measured by experiment. Whereas the replication of the original computed values for the gas-phase reaction proved entirely successful, several issues were discovered when a continuum solvent model was used. These included uncertainty regarding the parameters and methods used for the calculations and also the coordinates for the original reactant and transition structures, via their provision as data in the ESI. The original conclusions, in which a numerical mis-match between the magnitude of the computed and experimentally measured KIE was attributed to significant deviations from transition structure theory, are here instead rationalised as a manifestation of basis-set effects in the computation. Transition state theory appears to be operating successfully. We now recommend the use of basis sets of triple- or quadruple-ζ quality, rather than the split-valence level previously employed, that dispersion energy corrections be included and that a continuum solvent model using smoothed reaction cavities is essential for effective geometry optimisation and hence accurate normal coordinate analysis. An outlying experimental KIE obtained for chloroform as solvent is attributed to a small level of an explicit hydrogen bonded interaction with the substrate. A temperature outlier for the measured KIE at 195 K is suggested for further experimental investigation, although it may also be an indication of an unusually abrupt incursion of hydrogen tunnelling which would need non-Born–Oppenheimer methods in which nuclear quantum effects are included to be more accurately modelled. We predict KIEs for new substituents, of which those for R = NMe2 are significantly larger than those for R = H. This approach could be useful in designing variations of the Swern reagent that could lead to synthesis of aldehydes incorporating much higher levels of deuterium. The use of FAIR data rather than the traditional model of its inclusion in the ESI is discussed, and two data discovery tools exploiting these FAIR attributes are suggested.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
