Lucas W. Gauthier, Morgane Gossez, Christophe Malcus, Sébastien Viel, Guillaume Monneret, Remy Bordonné, Linda Pons, Sara Cabet, Marion Delous, Sylvie Mazoyer, Audrey Putoux, Patrick Edery
{"title":"双胞胎姐妹的 B 细胞免疫缺陷扩大了澳门巴黎人娱乐官网的表型。","authors":"Lucas W. Gauthier, Morgane Gossez, Christophe Malcus, Sébastien Viel, Guillaume Monneret, Remy Bordonné, Linda Pons, Sara Cabet, Marion Delous, Sylvie Mazoyer, Audrey Putoux, Patrick Edery","doi":"10.1111/cge.14571","DOIUrl":null,"url":null,"abstract":"<p>Microcephalic osteodysplastic primordial dwarfism type I (MOPDI) is a very rare and severe autosomal recessive disorder characterized by marked intrauterine growth retardation, skeletal dysplasia, microcephaly and brain malformations. MOPDI is caused by biallelic mutations in <i>RNU4ATAC</i>, a non-coding gene involved in U12-type splicing of 1% of the introns in the genome, which are recognized by their specific splicing consensus sequences. Here, we describe a unique observation of immunodeficiency in twin sisters with mild MOPDI, who harbor a novel n.108_126del mutation, encompassing part of the U4atac snRNA 3′ stem-loop and Sm protein binding site, and the previously reported n.111G>A mutation. Interestingly, both twin sisters show mild B-cell anomalies, including low naive B-cell counts and increased memory B-cell and plasmablasts counts, suggesting partial and transitory blockage of B-cell maturation and/or excessive activation of naive B-cells. Hence, the localization of a mutation in stem II of U4atac snRNA, as observed in another <i>RNU4ATAC</i>-opathy with immunodeficiency, that is, Roifman syndrome (RFMN), is not required for the occurrence of an immune deficiency. Finally, we emphasize the importance of considering immunodeficiency in MOPDI management to reduce the risk of serious infectious episodes.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":"106 4","pages":"476-482"},"PeriodicalIF":2.3000,"publicationDate":"2024-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14571","citationCount":"0","resultStr":"{\"title\":\"B-cell immune deficiency in twin sisters expands the phenotype of MOPDI\",\"authors\":\"Lucas W. Gauthier, Morgane Gossez, Christophe Malcus, Sébastien Viel, Guillaume Monneret, Remy Bordonné, Linda Pons, Sara Cabet, Marion Delous, Sylvie Mazoyer, Audrey Putoux, Patrick Edery\",\"doi\":\"10.1111/cge.14571\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Microcephalic osteodysplastic primordial dwarfism type I (MOPDI) is a very rare and severe autosomal recessive disorder characterized by marked intrauterine growth retardation, skeletal dysplasia, microcephaly and brain malformations. MOPDI is caused by biallelic mutations in <i>RNU4ATAC</i>, a non-coding gene involved in U12-type splicing of 1% of the introns in the genome, which are recognized by their specific splicing consensus sequences. Here, we describe a unique observation of immunodeficiency in twin sisters with mild MOPDI, who harbor a novel n.108_126del mutation, encompassing part of the U4atac snRNA 3′ stem-loop and Sm protein binding site, and the previously reported n.111G>A mutation. Interestingly, both twin sisters show mild B-cell anomalies, including low naive B-cell counts and increased memory B-cell and plasmablasts counts, suggesting partial and transitory blockage of B-cell maturation and/or excessive activation of naive B-cells. Hence, the localization of a mutation in stem II of U4atac snRNA, as observed in another <i>RNU4ATAC</i>-opathy with immunodeficiency, that is, Roifman syndrome (RFMN), is not required for the occurrence of an immune deficiency. Finally, we emphasize the importance of considering immunodeficiency in MOPDI management to reduce the risk of serious infectious episodes.</p>\",\"PeriodicalId\":10354,\"journal\":{\"name\":\"Clinical Genetics\",\"volume\":\"106 4\",\"pages\":\"476-482\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2024-06-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14571\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cge.14571\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14571","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
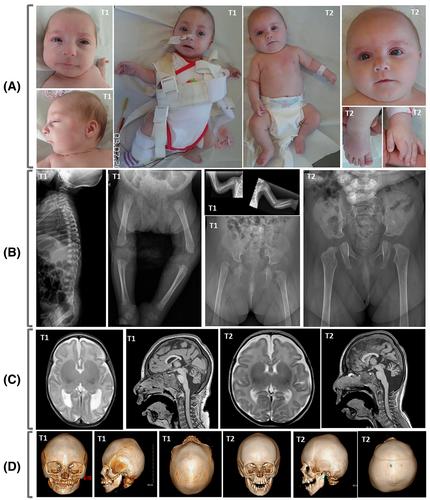
小头畸形骨发育不良性原始侏儒症 I 型(MOPDI)是一种非常罕见的严重常染色体隐性遗传疾病,以明显的宫内发育迟缓、骨骼发育不良、小头畸形和脑畸形为特征。RNU4ATAC 是一种非编码基因,参与基因组中 1%内含子的 U12 型剪接,这些内含子由其特定的剪接共识序列识别。在这里,我们描述了在一对患有轻度澳门巴黎人娱乐官网的孪生姐妹中观察到的免疫缺陷的独特现象,这对孪生姐妹携带一个新的n.108_126del突变,该突变包括U4atac snRNA 3'茎环和Sm蛋白结合位点的一部分,以及先前报道的n.111G>A突变。有趣的是,这对孪生姐妹都表现出轻微的 B 细胞异常,包括幼稚 B 细胞计数低、记忆 B 细胞和浆细胞计数增高,这表明 B 细胞成熟的部分和暂时性阻断和/或幼稚 B 细胞的过度激活。因此,U4atac snRNA干Ⅱ中的突变(如在另一种伴有免疫缺陷的RNU4ATAC病,即罗伊夫曼综合征(RFMN)中所观察到的那样)并不是发生免疫缺陷的必要条件。最后,我们强调在澳门巴黎人娱乐官网管理中考虑免疫缺陷的重要性,以降低严重感染发作的风险。
B-cell immune deficiency in twin sisters expands the phenotype of MOPDI
Microcephalic osteodysplastic primordial dwarfism type I (MOPDI) is a very rare and severe autosomal recessive disorder characterized by marked intrauterine growth retardation, skeletal dysplasia, microcephaly and brain malformations. MOPDI is caused by biallelic mutations in RNU4ATAC, a non-coding gene involved in U12-type splicing of 1% of the introns in the genome, which are recognized by their specific splicing consensus sequences. Here, we describe a unique observation of immunodeficiency in twin sisters with mild MOPDI, who harbor a novel n.108_126del mutation, encompassing part of the U4atac snRNA 3′ stem-loop and Sm protein binding site, and the previously reported n.111G>A mutation. Interestingly, both twin sisters show mild B-cell anomalies, including low naive B-cell counts and increased memory B-cell and plasmablasts counts, suggesting partial and transitory blockage of B-cell maturation and/or excessive activation of naive B-cells. Hence, the localization of a mutation in stem II of U4atac snRNA, as observed in another RNU4ATAC-opathy with immunodeficiency, that is, Roifman syndrome (RFMN), is not required for the occurrence of an immune deficiency. Finally, we emphasize the importance of considering immunodeficiency in MOPDI management to reduce the risk of serious infectious episodes.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
