{"title":"玻璃转化温度的分子量依赖性是由链端效应驱动的吗?","authors":"William F. Drayer, and , David S. Simmons*, ","doi":"10.1021/acs.macromol.4c00419","DOIUrl":null,"url":null,"abstract":"<p >The immense dependence of the glass transition temperature <i>T</i><sub>g</sub> on molecular weight <i>M</i> is one of the most fundamentally and practically important features of polymer glass formation. Here, we report on molecular dynamics simulations of three model linear polymers of substantially different complexity demonstrating that the 70-year-old canonical explanation of this dependence (a simple chain end dilution effect) is likely incorrect at leading order. Our data show that end effects are present only in relatively stiff polymers and, furthermore, that the magnitude of these end effects diminish on cooling. We find that <i>T</i><sub>g</sub>(<i>M</i>) trends are instead dominated by shifts in <i>T</i><sub>g</sub> throughout the entire polymer chain rather than through a chain end effect. We show that these data can be rationalized via a generic two-barrier model of <i>T</i><sub>g</sub> and its <i>M</i>-dependence, motivated by the Elastically Collective Nonlinear Langevin Equation theory. More broadly, this work indicates need to reopen the question of the origin of the <i>T</i><sub>g</sub>(<i>M</i>) dependence in linear polymers (and macromolecules at large), as well as an opportunity to reveal new glass formation physics with renewed study of <i>M</i> effects on <i>T</i><sub>g</sub>.</p>","PeriodicalId":51,"journal":{"name":"Macromolecules","volume":"57 12","pages":"5589–5597"},"PeriodicalIF":5.2000,"publicationDate":"2024-06-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Is the Molecular Weight Dependence of the Glass Transition Temperature Driven by a Chain End Effect?\",\"authors\":\"William F. Drayer, and , David S. Simmons*, \",\"doi\":\"10.1021/acs.macromol.4c00419\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The immense dependence of the glass transition temperature <i>T</i><sub>g</sub> on molecular weight <i>M</i> is one of the most fundamentally and practically important features of polymer glass formation. Here, we report on molecular dynamics simulations of three model linear polymers of substantially different complexity demonstrating that the 70-year-old canonical explanation of this dependence (a simple chain end dilution effect) is likely incorrect at leading order. Our data show that end effects are present only in relatively stiff polymers and, furthermore, that the magnitude of these end effects diminish on cooling. We find that <i>T</i><sub>g</sub>(<i>M</i>) trends are instead dominated by shifts in <i>T</i><sub>g</sub> throughout the entire polymer chain rather than through a chain end effect. We show that these data can be rationalized via a generic two-barrier model of <i>T</i><sub>g</sub> and its <i>M</i>-dependence, motivated by the Elastically Collective Nonlinear Langevin Equation theory. More broadly, this work indicates need to reopen the question of the origin of the <i>T</i><sub>g</sub>(<i>M</i>) dependence in linear polymers (and macromolecules at large), as well as an opportunity to reveal new glass formation physics with renewed study of <i>M</i> effects on <i>T</i><sub>g</sub>.</p>\",\"PeriodicalId\":51,\"journal\":{\"name\":\"Macromolecules\",\"volume\":\"57 12\",\"pages\":\"5589–5597\"},\"PeriodicalIF\":5.2000,\"publicationDate\":\"2024-06-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Macromolecules\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.macromol.4c00419\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"POLYMER SCIENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Macromolecules","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.macromol.4c00419","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"POLYMER SCIENCE","Score":null,"Total":0}
引用次数: 0
摘要
玻璃化转变温度 Tg 与分子量 M 的巨大相关性是聚合物玻璃化形成过程中最重要的基本特征之一。在此,我们报告了对三种复杂程度大相径庭的线性聚合物模型的分子动力学模拟,结果表明,对这种依赖性已有 70 年历史的经典解释(简单的链端稀释效应)在前阶很可能是不正确的。我们的数据显示,只有在相对较硬的聚合物中才会出现末端效应,而且这些末端效应的程度会随着冷却而减弱。我们发现,Tg(M)的变化趋势是由整个聚合物链的 Tg 变化主导的,而不是通过链端效应。我们通过弹性集合非线性朗格文方程理论提出的 Tg 及其 M 依赖性的通用双屏障模型,证明这些数据是合理的。从更广泛的意义上讲,这项研究表明有必要重新探讨线性聚合物(以及整个高分子)的 Tg(M)依赖性的起源问题,同时也为重新研究 M 对 Tg 的影响提供了揭示新的玻璃形成物理学的机会。
Is the Molecular Weight Dependence of the Glass Transition Temperature Driven by a Chain End Effect?
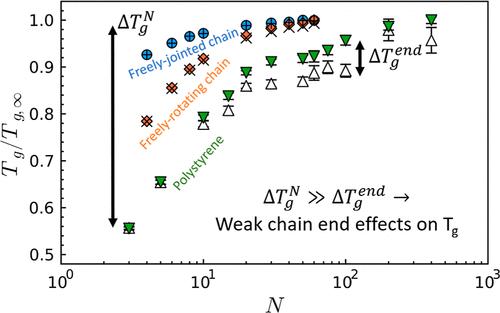
The immense dependence of the glass transition temperature Tg on molecular weight M is one of the most fundamentally and practically important features of polymer glass formation. Here, we report on molecular dynamics simulations of three model linear polymers of substantially different complexity demonstrating that the 70-year-old canonical explanation of this dependence (a simple chain end dilution effect) is likely incorrect at leading order. Our data show that end effects are present only in relatively stiff polymers and, furthermore, that the magnitude of these end effects diminish on cooling. We find that Tg(M) trends are instead dominated by shifts in Tg throughout the entire polymer chain rather than through a chain end effect. We show that these data can be rationalized via a generic two-barrier model of Tg and its M-dependence, motivated by the Elastically Collective Nonlinear Langevin Equation theory. More broadly, this work indicates need to reopen the question of the origin of the Tg(M) dependence in linear polymers (and macromolecules at large), as well as an opportunity to reveal new glass formation physics with renewed study of M effects on Tg.
期刊介绍:
Macromolecules publishes original, fundamental, and impactful research on all aspects of polymer science. Topics of interest include synthesis (e.g., controlled polymerizations, polymerization catalysis, post polymerization modification, new monomer structures and polymer architectures, and polymerization mechanisms/kinetics analysis); phase behavior, thermodynamics, dynamic, and ordering/disordering phenomena (e.g., self-assembly, gelation, crystallization, solution/melt/solid-state characteristics); structure and properties (e.g., mechanical and rheological properties, surface/interfacial characteristics, electronic and transport properties); new state of the art characterization (e.g., spectroscopy, scattering, microscopy, rheology), simulation (e.g., Monte Carlo, molecular dynamics, multi-scale/coarse-grained modeling), and theoretical methods. Renewable/sustainable polymers, polymer networks, responsive polymers, electro-, magneto- and opto-active macromolecules, inorganic polymers, charge-transporting polymers (ion-containing, semiconducting, and conducting), nanostructured polymers, and polymer composites are also of interest. Typical papers published in Macromolecules showcase important and innovative concepts, experimental methods/observations, and theoretical/computational approaches that demonstrate a fundamental advance in the understanding of polymers.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
