Amar Y. Al-Ansi, Gamal H. Al-Shawesh, Xiao Ru* and Zijing Lin*,
{"title":"基于量子力学的快速可靠的结合姿态结构预测。","authors":"Amar Y. Al-Ansi, Gamal H. Al-Shawesh, Xiao Ru* and Zijing Lin*, ","doi":"10.1021/acs.jpcb.4c02596","DOIUrl":null,"url":null,"abstract":"<p >Predicting the binding poses of docking with an accurate estimation of binding energies is highly important but very challenging in computational drug design. A quantum mechanics (QM) calculation-based docking approach considering multiple conformations and orientations of the ligand is introduced here to tackle the problem. This QM docking consists of three steps: generating an ensemble of binding poses with a conventional docking simulation, computing the binding energies with self-consistent charge density functional theory tightly binding with dispersion correction (DFTB-D) to selecting the 10 top binding modes, and optimizing the selected binding mode structures using the ONIOM(DFTB:PM7) technique to determine the binding poses. The ONIOM(DFTB-D:PM6) docking approach is tested on 121 ligand–receptor biocomplexes with the crystal structures obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). The result shows that the new method is highly satisfactory for the accurate prediction of the binding poses. The new docking method should be beneficial to structure-based drug design.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":"128 25","pages":"6059–6070"},"PeriodicalIF":2.9000,"publicationDate":"2024-06-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Quantum Mechanics-Based Fast and Reliable Prediction of Binding Pose Structures\",\"authors\":\"Amar Y. Al-Ansi, Gamal H. Al-Shawesh, Xiao Ru* and Zijing Lin*, \",\"doi\":\"10.1021/acs.jpcb.4c02596\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Predicting the binding poses of docking with an accurate estimation of binding energies is highly important but very challenging in computational drug design. A quantum mechanics (QM) calculation-based docking approach considering multiple conformations and orientations of the ligand is introduced here to tackle the problem. This QM docking consists of three steps: generating an ensemble of binding poses with a conventional docking simulation, computing the binding energies with self-consistent charge density functional theory tightly binding with dispersion correction (DFTB-D) to selecting the 10 top binding modes, and optimizing the selected binding mode structures using the ONIOM(DFTB:PM7) technique to determine the binding poses. The ONIOM(DFTB-D:PM6) docking approach is tested on 121 ligand–receptor biocomplexes with the crystal structures obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). The result shows that the new method is highly satisfactory for the accurate prediction of the binding poses. The new docking method should be beneficial to structure-based drug design.</p>\",\"PeriodicalId\":60,\"journal\":{\"name\":\"The Journal of Physical Chemistry B\",\"volume\":\"128 25\",\"pages\":\"6059–6070\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-06-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry B\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcb.4c02596\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcb.4c02596","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Quantum Mechanics-Based Fast and Reliable Prediction of Binding Pose Structures
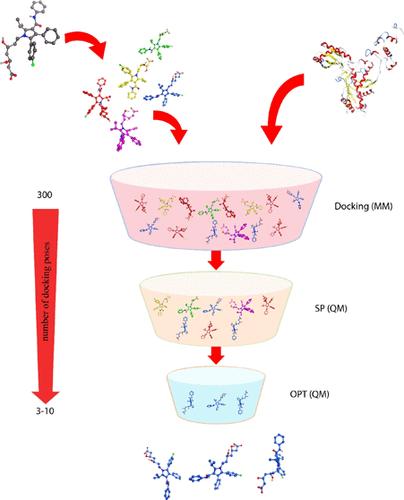
Predicting the binding poses of docking with an accurate estimation of binding energies is highly important but very challenging in computational drug design. A quantum mechanics (QM) calculation-based docking approach considering multiple conformations and orientations of the ligand is introduced here to tackle the problem. This QM docking consists of three steps: generating an ensemble of binding poses with a conventional docking simulation, computing the binding energies with self-consistent charge density functional theory tightly binding with dispersion correction (DFTB-D) to selecting the 10 top binding modes, and optimizing the selected binding mode structures using the ONIOM(DFTB:PM7) technique to determine the binding poses. The ONIOM(DFTB-D:PM6) docking approach is tested on 121 ligand–receptor biocomplexes with the crystal structures obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). The result shows that the new method is highly satisfactory for the accurate prediction of the binding poses. The new docking method should be beneficial to structure-based drug design.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
