Friederike Rathing, Dirk Schepmann, Bernhard Wünsch
{"title":"酚类 GluN2B 选择性 NMDA 受体拮抗剂的醌类生物异斯特。","authors":"Friederike Rathing, Dirk Schepmann, Bernhard Wünsch","doi":"10.1002/ardp.202400279","DOIUrl":null,"url":null,"abstract":"<p>Cyclopenta[<i>g</i>]quinolones of type <b>4</b> were designed with the aim to bioisosterically replace the phenol of potent GluN2B ligands such as ifenprodil and Ro 25-6981 by the quinolone system and to restrict the conformational flexibility of the aminopropanol substructure in a cyclopentane system. The designed ligands were synthesized in an eight-step sequence starting with terephthalaldehyde (<b>5</b>). Key steps pf the synthesis were the intramolecular Friedel–Crafts acylation of propionic acids <b>10</b> to yield the cyclopenta[<i>g</i>]quinolinediones <b>11</b> and the Mannich reaction of diketone <b>11a</b> followed by conjugate addition at the α,β-unsaturated ketone <b>12a</b>. Although the quinolones <b>13a</b>, <b>15a</b>, and <b>16a</b> contain an H-bond donor group (secondary lactam) as ifenprodil and Ro 25-6981, they show only moderate GluN2B affinity (<i>K</i><sub>i</sub> > 410 nM). However, the introduction of lipophilic substituents at the quinolone N-atom resulted in more than 10-fold increased GluN2B affinity of the benzyl and benzyloxymethyl derivatives <i>cis</i>-<b>13c</b> (<i>K</i><sub>o</sub> = 36 nM) and <b>13e</b> (<i>K</i><sub>o</sub> = 27 nM). All compounds are selective over the phencyclidine (PCP) binding site of the <i>N</i>-methyl-<i>D</i>-aspartate (NMDA) receptor. The benzyl derivative <b>13c</b> showed six- and threefold selectivity over σ<sub>1</sub> and σ<sub>2</sub> receptors, respectively.</p>","PeriodicalId":128,"journal":{"name":"Archiv der Pharmazie","volume":"357 9","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ardp.202400279","citationCount":"0","resultStr":"{\"title\":\"Quinolone bioisosteres of phenolic GluN2B-selective NMDA receptor antagonists\",\"authors\":\"Friederike Rathing, Dirk Schepmann, Bernhard Wünsch\",\"doi\":\"10.1002/ardp.202400279\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Cyclopenta[<i>g</i>]quinolones of type <b>4</b> were designed with the aim to bioisosterically replace the phenol of potent GluN2B ligands such as ifenprodil and Ro 25-6981 by the quinolone system and to restrict the conformational flexibility of the aminopropanol substructure in a cyclopentane system. The designed ligands were synthesized in an eight-step sequence starting with terephthalaldehyde (<b>5</b>). Key steps pf the synthesis were the intramolecular Friedel–Crafts acylation of propionic acids <b>10</b> to yield the cyclopenta[<i>g</i>]quinolinediones <b>11</b> and the Mannich reaction of diketone <b>11a</b> followed by conjugate addition at the α,β-unsaturated ketone <b>12a</b>. Although the quinolones <b>13a</b>, <b>15a</b>, and <b>16a</b> contain an H-bond donor group (secondary lactam) as ifenprodil and Ro 25-6981, they show only moderate GluN2B affinity (<i>K</i><sub>i</sub> > 410 nM). However, the introduction of lipophilic substituents at the quinolone N-atom resulted in more than 10-fold increased GluN2B affinity of the benzyl and benzyloxymethyl derivatives <i>cis</i>-<b>13c</b> (<i>K</i><sub>o</sub> = 36 nM) and <b>13e</b> (<i>K</i><sub>o</sub> = 27 nM). All compounds are selective over the phencyclidine (PCP) binding site of the <i>N</i>-methyl-<i>D</i>-aspartate (NMDA) receptor. The benzyl derivative <b>13c</b> showed six- and threefold selectivity over σ<sub>1</sub> and σ<sub>2</sub> receptors, respectively.</p>\",\"PeriodicalId\":128,\"journal\":{\"name\":\"Archiv der Pharmazie\",\"volume\":\"357 9\",\"pages\":\"\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ardp.202400279\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Archiv der Pharmazie\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ardp.202400279\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archiv der Pharmazie","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ardp.202400279","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Quinolone bioisosteres of phenolic GluN2B-selective NMDA receptor antagonists
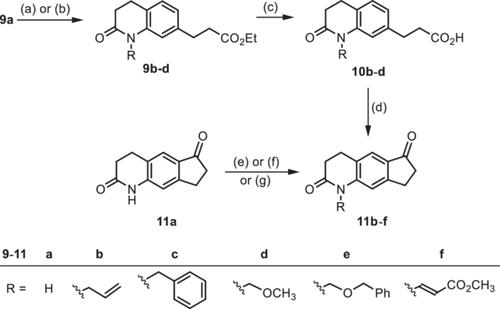
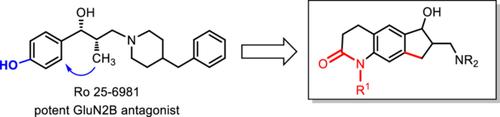
Cyclopenta[g]quinolones of type 4 were designed with the aim to bioisosterically replace the phenol of potent GluN2B ligands such as ifenprodil and Ro 25-6981 by the quinolone system and to restrict the conformational flexibility of the aminopropanol substructure in a cyclopentane system. The designed ligands were synthesized in an eight-step sequence starting with terephthalaldehyde (5). Key steps pf the synthesis were the intramolecular Friedel–Crafts acylation of propionic acids 10 to yield the cyclopenta[g]quinolinediones 11 and the Mannich reaction of diketone 11a followed by conjugate addition at the α,β-unsaturated ketone 12a. Although the quinolones 13a, 15a, and 16a contain an H-bond donor group (secondary lactam) as ifenprodil and Ro 25-6981, they show only moderate GluN2B affinity (Ki > 410 nM). However, the introduction of lipophilic substituents at the quinolone N-atom resulted in more than 10-fold increased GluN2B affinity of the benzyl and benzyloxymethyl derivatives cis-13c (Ko = 36 nM) and 13e (Ko = 27 nM). All compounds are selective over the phencyclidine (PCP) binding site of the N-methyl-D-aspartate (NMDA) receptor. The benzyl derivative 13c showed six- and threefold selectivity over σ1 and σ2 receptors, respectively.
期刊介绍:
Archiv der Pharmazie - Chemistry in Life Sciences is an international journal devoted to research and development in all fields of pharmaceutical and medicinal chemistry. Emphasis is put on papers combining synthetic organic chemistry, structural biology, molecular modelling, bioorganic chemistry, natural products chemistry, biochemistry or analytical methods with pharmaceutical or medicinal aspects such as biological activity. The focus of this journal is put on original research papers, but other scientifically valuable contributions (e.g. reviews, minireviews, highlights, symposia contributions, discussions, and essays) are also welcome.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
