{"title":"对确诊为心脏通道病的家族进行全基因组测序,发现了全外显子测序所遗漏的结构变异。","authors":"Vigneshwar Senthivel, Bani Jolly, Arvinden VR, Anjali Bajaj, Rahul Bhoyar, Mohamed Imran, Harie Vignesh, Mohit Kumar Divakar, Gautam Sharma, Nitin Rai, Kapil Kumar, Jayakrishnan MP, Maniram Krishna, Jeyaprakash Shenthar, Muzaffar Ali, Shaad Abqari, Gulnaz Nadri, Vinod Scaria, Nitish Naik, Sridhar Sivasubbu","doi":"10.1038/s10038-024-01265-2","DOIUrl":null,"url":null,"abstract":"Cardiac channelopathies are a group of heritable disorders that affect the heart’s electrical activity due to genetic variations present in genes coding for ion channels. With the advent of new sequencing technologies, molecular diagnosis of these disorders in patients has paved the way for early identification, therapeutic management and family screening. The objective of this retrospective study was to understand the efficacy of whole-genome sequencing in diagnosing patients with suspected cardiac channelopathies who were reported negative after whole exome sequencing and analysis. We employed a 3-tier analysis approach to identify nonsynonymous variations and loss-of-function variations missed by exome sequencing, and structural variations that are better resolved only by sequencing whole genomes. By performing whole genome sequencing and analyzing 25 exome-negative cardiac channelopathy patients, we identified 3 pathogenic variations. These include a heterozygous likely pathogenic nonsynonymous variation, CACNA1C:NM_000719:exon19:c.C2570G:p. P857R, which causes autosomal dominant long QT syndrome in the absence of Timothy syndrome, a heterozygous loss-of-function variation CASQ2:NM_001232.4:c.420+2T>C classified as pathogenic, and a 9.2 kb structural variation that spans exon 2 of the KCNQ1 gene, which is likely to cause Jervell-Lange-Nielssen syndrome. In addition, we also identified a loss-of-function variation and 16 structural variations of unknown significance (VUS). Further studies are required to elucidate the role of these identified VUS in gene regulation and decipher the underlying genetic and molecular mechanisms of these disorders. Our present study serves as a pilot for understanding the utility of WGS over clinical exomes in diagnosing cardiac channelopathy disorders.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 9","pages":"455-465"},"PeriodicalIF":2.3000,"publicationDate":"2024-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Whole genome sequencing of families diagnosed with cardiac channelopathies reveals structural variants missed by whole exome sequencing\",\"authors\":\"Vigneshwar Senthivel, Bani Jolly, Arvinden VR, Anjali Bajaj, Rahul Bhoyar, Mohamed Imran, Harie Vignesh, Mohit Kumar Divakar, Gautam Sharma, Nitin Rai, Kapil Kumar, Jayakrishnan MP, Maniram Krishna, Jeyaprakash Shenthar, Muzaffar Ali, Shaad Abqari, Gulnaz Nadri, Vinod Scaria, Nitish Naik, Sridhar Sivasubbu\",\"doi\":\"10.1038/s10038-024-01265-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Cardiac channelopathies are a group of heritable disorders that affect the heart’s electrical activity due to genetic variations present in genes coding for ion channels. With the advent of new sequencing technologies, molecular diagnosis of these disorders in patients has paved the way for early identification, therapeutic management and family screening. The objective of this retrospective study was to understand the efficacy of whole-genome sequencing in diagnosing patients with suspected cardiac channelopathies who were reported negative after whole exome sequencing and analysis. We employed a 3-tier analysis approach to identify nonsynonymous variations and loss-of-function variations missed by exome sequencing, and structural variations that are better resolved only by sequencing whole genomes. By performing whole genome sequencing and analyzing 25 exome-negative cardiac channelopathy patients, we identified 3 pathogenic variations. These include a heterozygous likely pathogenic nonsynonymous variation, CACNA1C:NM_000719:exon19:c.C2570G:p. P857R, which causes autosomal dominant long QT syndrome in the absence of Timothy syndrome, a heterozygous loss-of-function variation CASQ2:NM_001232.4:c.420+2T>C classified as pathogenic, and a 9.2 kb structural variation that spans exon 2 of the KCNQ1 gene, which is likely to cause Jervell-Lange-Nielssen syndrome. In addition, we also identified a loss-of-function variation and 16 structural variations of unknown significance (VUS). Further studies are required to elucidate the role of these identified VUS in gene regulation and decipher the underlying genetic and molecular mechanisms of these disorders. Our present study serves as a pilot for understanding the utility of WGS over clinical exomes in diagnosing cardiac channelopathy disorders.\",\"PeriodicalId\":16077,\"journal\":{\"name\":\"Journal of Human Genetics\",\"volume\":\"69 9\",\"pages\":\"455-465\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2024-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s10038-024-01265-2\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01265-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Whole genome sequencing of families diagnosed with cardiac channelopathies reveals structural variants missed by whole exome sequencing
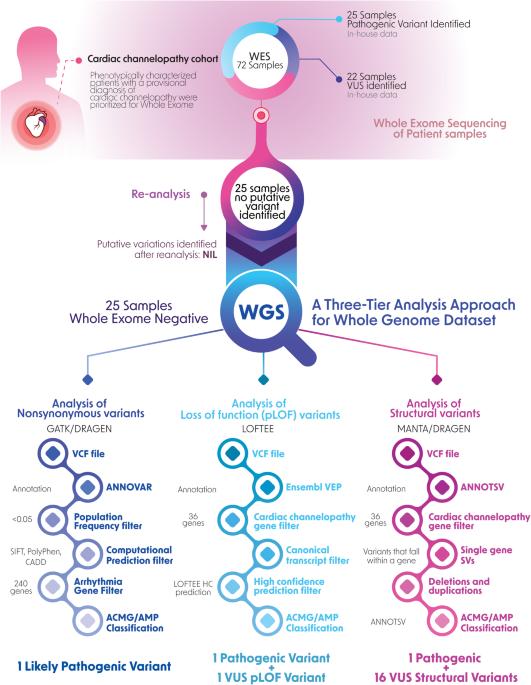
Cardiac channelopathies are a group of heritable disorders that affect the heart’s electrical activity due to genetic variations present in genes coding for ion channels. With the advent of new sequencing technologies, molecular diagnosis of these disorders in patients has paved the way for early identification, therapeutic management and family screening. The objective of this retrospective study was to understand the efficacy of whole-genome sequencing in diagnosing patients with suspected cardiac channelopathies who were reported negative after whole exome sequencing and analysis. We employed a 3-tier analysis approach to identify nonsynonymous variations and loss-of-function variations missed by exome sequencing, and structural variations that are better resolved only by sequencing whole genomes. By performing whole genome sequencing and analyzing 25 exome-negative cardiac channelopathy patients, we identified 3 pathogenic variations. These include a heterozygous likely pathogenic nonsynonymous variation, CACNA1C:NM_000719:exon19:c.C2570G:p. P857R, which causes autosomal dominant long QT syndrome in the absence of Timothy syndrome, a heterozygous loss-of-function variation CASQ2:NM_001232.4:c.420+2T>C classified as pathogenic, and a 9.2 kb structural variation that spans exon 2 of the KCNQ1 gene, which is likely to cause Jervell-Lange-Nielssen syndrome. In addition, we also identified a loss-of-function variation and 16 structural variations of unknown significance (VUS). Further studies are required to elucidate the role of these identified VUS in gene regulation and decipher the underlying genetic and molecular mechanisms of these disorders. Our present study serves as a pilot for understanding the utility of WGS over clinical exomes in diagnosing cardiac channelopathy disorders.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
