Marta Gałyńska, Matheus Morato F. de Moraes, Paweł Tecmer and Katharina Boguslawski
{"title":"探究钼辅助因子的催化机理:新型耦合簇研究","authors":"Marta Gałyńska, Matheus Morato F. de Moraes, Paweł Tecmer and Katharina Boguslawski","doi":"10.1039/D4CP01500B","DOIUrl":null,"url":null,"abstract":"<p >In this work, we use modern electronic structure methods to model the catalytic mechanism of different variants of the molybdenum cofactor (Moco). We investigate the dependence of various Moco model systems on structural relaxation and the importance of environmental effects for five critical points along the reaction coordinate with the DMSO and NO<small><sub>3</sub></small><small><sup>−</sup></small> substrates. Furthermore, we scrutinize the performance of various coupled-cluster approaches for modeling the relative energies along the investigated reaction paths, focusing on several pair coupled cluster doubles (pCCD) flavors and conventional coupled cluster approximations. Moreover, we elucidate the Mo–O bond formation using orbital-based quantum information measures, which highlight the flow of <em>σ</em><small><sub>M–O</sub></small> bond formation and <em>σ</em><small><sub>N/S–O</sub></small> bond breaking. Our study shows that pCCD-based models are a viable alternative to conventional methods and offer us unique insights into the bonding situation along a reaction coordinate. Finally, this work highlights the importance of environmental effects or changes in the core and, consequently, in the model itself to elucidate the change in activity of different Moco variants.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 27","pages":" 18918-18929"},"PeriodicalIF":3.0000,"publicationDate":"2024-06-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Delving into the catalytic mechanism of molybdenum cofactors: a novel coupled cluster study†\",\"authors\":\"Marta Gałyńska, Matheus Morato F. de Moraes, Paweł Tecmer and Katharina Boguslawski\",\"doi\":\"10.1039/D4CP01500B\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In this work, we use modern electronic structure methods to model the catalytic mechanism of different variants of the molybdenum cofactor (Moco). We investigate the dependence of various Moco model systems on structural relaxation and the importance of environmental effects for five critical points along the reaction coordinate with the DMSO and NO<small><sub>3</sub></small><small><sup>−</sup></small> substrates. Furthermore, we scrutinize the performance of various coupled-cluster approaches for modeling the relative energies along the investigated reaction paths, focusing on several pair coupled cluster doubles (pCCD) flavors and conventional coupled cluster approximations. Moreover, we elucidate the Mo–O bond formation using orbital-based quantum information measures, which highlight the flow of <em>σ</em><small><sub>M–O</sub></small> bond formation and <em>σ</em><small><sub>N/S–O</sub></small> bond breaking. Our study shows that pCCD-based models are a viable alternative to conventional methods and offer us unique insights into the bonding situation along a reaction coordinate. Finally, this work highlights the importance of environmental effects or changes in the core and, consequently, in the model itself to elucidate the change in activity of different Moco variants.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 27\",\"pages\":\" 18918-18929\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-06-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01500b\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01500b","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Delving into the catalytic mechanism of molybdenum cofactors: a novel coupled cluster study†
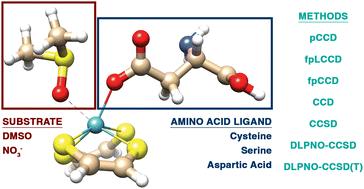
In this work, we use modern electronic structure methods to model the catalytic mechanism of different variants of the molybdenum cofactor (Moco). We investigate the dependence of various Moco model systems on structural relaxation and the importance of environmental effects for five critical points along the reaction coordinate with the DMSO and NO3− substrates. Furthermore, we scrutinize the performance of various coupled-cluster approaches for modeling the relative energies along the investigated reaction paths, focusing on several pair coupled cluster doubles (pCCD) flavors and conventional coupled cluster approximations. Moreover, we elucidate the Mo–O bond formation using orbital-based quantum information measures, which highlight the flow of σM–O bond formation and σN/S–O bond breaking. Our study shows that pCCD-based models are a viable alternative to conventional methods and offer us unique insights into the bonding situation along a reaction coordinate. Finally, this work highlights the importance of environmental effects or changes in the core and, consequently, in the model itself to elucidate the change in activity of different Moco variants.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
