{"title":"通过催化α-氰基二氮羰基化合物的炔环丙烷化反应获得环己二烯和苯并呋喃衍生物。","authors":"","doi":"10.1039/d4ob00696h","DOIUrl":null,"url":null,"abstract":"<div><p>The arene cyclopropanation between diazo compounds and benzene is well known to produce a tautomeric mixture of norcaradiene and cycloheptatriene in favour of the latter species. Nevertheless, previous studies have suggested that the initially formed norcaradiene can be stabilized by a C-7 cyano group with prevention of its 6π-electrocyclic ring opening. According to this feature, a synthetic route to functionalized cyclohexadienes has been designed using α-cyanodiazoacetates and α-diazo-β-ketonitriles as the starting materials, respectively. The Rh<sub>2</sub>(esp)<sub>2</sub>-catalyzed arene cyclopropanation of α-cyanodiazoacetates in benzene afforded the expected 7-alkoxycarbonyl-7-cyanonorcaradienes as isolable compounds, which then served as templates for the second cyclopropanation with ethyl diazoacetate or α-cyanodiazocarbonyls to enable the formation of bis(cyclopropanated) adducts. Their subsequent treatment with SmI<sub>2</sub> triggered a double ring-opening process, allowing for the generation of 1,4- and/or 1,3-cyclohexadienes as either regio- or diastereomeric mixtures. On the other hand, the norcaradienes generated from phenyl- or methyl-substituted α-diazo-β-ketonitriles were found to undergo an <em>in situ</em> rearrangement to yield dihydrobenzofurans that could be converted to benzofuran derivatives by DDQ oxidation.</p></div>","PeriodicalId":96,"journal":{"name":"Organic & Biomolecular Chemistry","volume":"22 27","pages":"Pages 5552-5560"},"PeriodicalIF":2.8000,"publicationDate":"2024-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Access to cyclohexadiene and benzofuran derivatives via catalytic arene cyclopropanation of α-cyanodiazocarbonyl compounds†\",\"authors\":\"\",\"doi\":\"10.1039/d4ob00696h\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The arene cyclopropanation between diazo compounds and benzene is well known to produce a tautomeric mixture of norcaradiene and cycloheptatriene in favour of the latter species. Nevertheless, previous studies have suggested that the initially formed norcaradiene can be stabilized by a C-7 cyano group with prevention of its 6π-electrocyclic ring opening. According to this feature, a synthetic route to functionalized cyclohexadienes has been designed using α-cyanodiazoacetates and α-diazo-β-ketonitriles as the starting materials, respectively. The Rh<sub>2</sub>(esp)<sub>2</sub>-catalyzed arene cyclopropanation of α-cyanodiazoacetates in benzene afforded the expected 7-alkoxycarbonyl-7-cyanonorcaradienes as isolable compounds, which then served as templates for the second cyclopropanation with ethyl diazoacetate or α-cyanodiazocarbonyls to enable the formation of bis(cyclopropanated) adducts. Their subsequent treatment with SmI<sub>2</sub> triggered a double ring-opening process, allowing for the generation of 1,4- and/or 1,3-cyclohexadienes as either regio- or diastereomeric mixtures. On the other hand, the norcaradienes generated from phenyl- or methyl-substituted α-diazo-β-ketonitriles were found to undergo an <em>in situ</em> rearrangement to yield dihydrobenzofurans that could be converted to benzofuran derivatives by DDQ oxidation.</p></div>\",\"PeriodicalId\":96,\"journal\":{\"name\":\"Organic & Biomolecular Chemistry\",\"volume\":\"22 27\",\"pages\":\"Pages 5552-5560\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-07-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Organic & Biomolecular Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/org/science/article/pii/S1477052024005627\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/6/12 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, ORGANIC\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Organic & Biomolecular Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/org/science/article/pii/S1477052024005627","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/12 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
Access to cyclohexadiene and benzofuran derivatives via catalytic arene cyclopropanation of α-cyanodiazocarbonyl compounds†
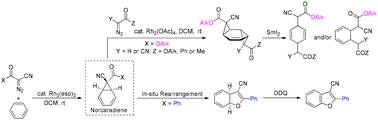
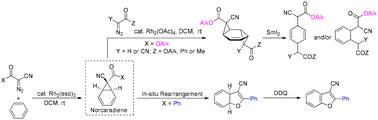
The arene cyclopropanation between diazo compounds and benzene is well known to produce a tautomeric mixture of norcaradiene and cycloheptatriene in favour of the latter species. Nevertheless, previous studies have suggested that the initially formed norcaradiene can be stabilized by a C-7 cyano group with prevention of its 6π-electrocyclic ring opening. According to this feature, a synthetic route to functionalized cyclohexadienes has been designed using α-cyanodiazoacetates and α-diazo-β-ketonitriles as the starting materials, respectively. The Rh2(esp)2-catalyzed arene cyclopropanation of α-cyanodiazoacetates in benzene afforded the expected 7-alkoxycarbonyl-7-cyanonorcaradienes as isolable compounds, which then served as templates for the second cyclopropanation with ethyl diazoacetate or α-cyanodiazocarbonyls to enable the formation of bis(cyclopropanated) adducts. Their subsequent treatment with SmI2 triggered a double ring-opening process, allowing for the generation of 1,4- and/or 1,3-cyclohexadienes as either regio- or diastereomeric mixtures. On the other hand, the norcaradienes generated from phenyl- or methyl-substituted α-diazo-β-ketonitriles were found to undergo an in situ rearrangement to yield dihydrobenzofurans that could be converted to benzofuran derivatives by DDQ oxidation.
期刊介绍:
Organic & Biomolecular Chemistry is an international journal using integrated research in chemistry-organic chemistry. Founded in 2003 by the Royal Society of Chemistry, the journal is published in Semimonthly issues and has been indexed by SCIE, a leading international database. The journal focuses on the key research and cutting-edge progress in the field of chemistry-organic chemistry, publishes and reports the research results in this field in a timely manner, and is committed to becoming a window and platform for rapid academic exchanges among peers in this field. The journal's impact factor in 2023 is 2.9, and its CiteScore is 5.5.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
